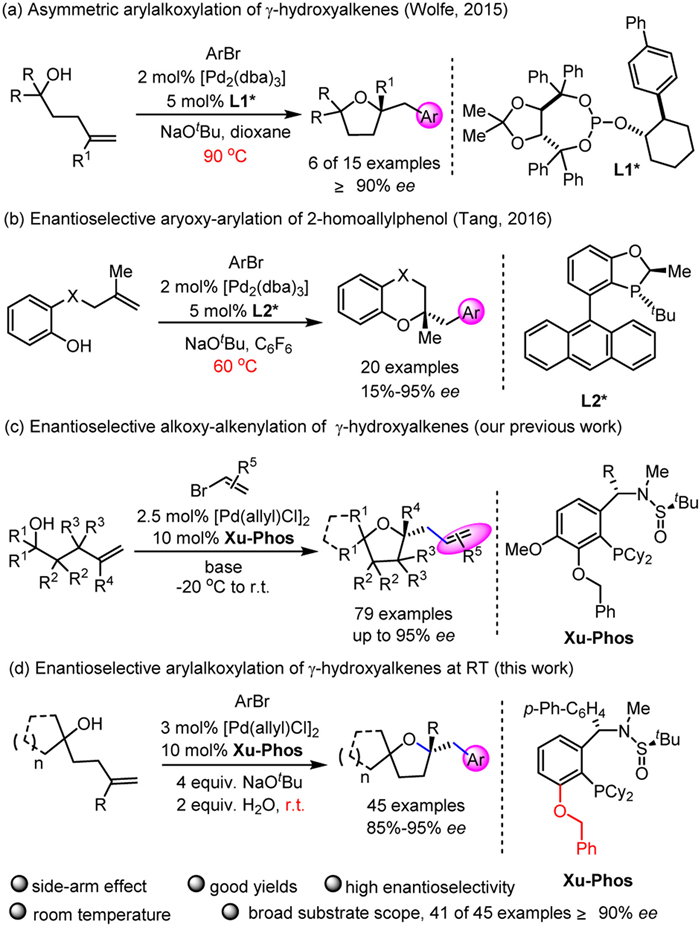
Scheme 1.
Pd-catalyzed enantioselective alkene carboetherification reactions for synthesis of chiral O-heterocycles.
Palladium/Xu-Phos-catalyzed enantioselective arylalkoxylation reaction of γ-hydroxyalkenes at room temperature
Shuai Zhu , Mingjie Chen , Haichao Shen , Hanming Ding , Wenbo Li , Junliang Zhang

Alkenes regarding as fundamental and readily available compounds, are useful building blocks in synthetic organic chemistry [1–13]. The difunctionalization of unactivated alkenes have been utilized as a powerful method to construct two distinct substituents on the two carbon atoms of the alkene moiety in one step, which has attracted increasing attention in academia and industry due to their low cost and high efficiency [14–16]. In the past several decades, inter/intramolecular difunctionalization of alkenes tethered a heteroatom-based nucleophile is particularly attractive, which offers various oxygen and nitrogen heterocycles (e.g., tetrahydrofuran, tetrahydropyran, lactone, pyrrolidine, and oxazine) [17–28]. Among them, hydroxy alkenes as important synthons have been disclosed to achieve hydroalkoxylation, haloalkoxylation, oxytrifluoromethylation, alkoxyalkenylation, and arylalkoxylation. In contrast to enantioselective hydroetherification and haloetherification reactions of alkenes [29–49], few examples have focused on the enantioselective carboalkoxylation reactions of alkenes, which can install C—O and C—C bonds at the same time. For instance, Chemler [50,51] and co-workers disclosed the enantioselective copper-catalyzed intramolecular carboetherification reactions of 4-pentenols with the use of box ligands. However, these processes demanded large amounts of oxidants and harsh conditions.
Powerful synthetic access has enabled arylation strategies incorporation into numerous efforts. As early as 2004, Wolfe's group uncovered the palladium-catalyzed arylalkoxylation of readily available γ-hydroxyalkenes with aryl halides to access the cascade cyclization/coupling products [52]. Until 2016, an enantioselective variant was achieved by using a new TADDOL/2-arylcyclohexanol-derived chiral phosphite ligand L1* (Scheme 1a) [53]. However, the reaction was carried out at high temperature (90 ℃) and in only six examples ee could be obtained with ≥90%. After that, Tang [54] and co-workers successfully developed a new AntPhos-derived ligand L2* for furnishing benzooxazines and chromans containing a quaternary stereocenter via Pd-catalyzed alkene aryloxyarylation reaction (Scheme 1b).
Owing to the great potential, the development of efficient processes to optical active 2,2′-multisubstituted tetrahydrofurans under mild conditions with a wild substrate scope is highly desired. Recently, we have reported a Pd/Xu-Phos-catalyzed enantioselective alkoxy-alkenylation of γ-hydroxyalkenes with alkenyl halides, delivering various chiral tetrahydrofurans in good yields with up to 95% ee (Scheme 1c) [55]. Inspired by the work of Wolfe, Tang and our group [56–66], a growing interest in the cascade alkoxylation/coupling reaction with aryl bromide has emerged. However, there are some challenges, such as harsh conditions, relatively poor reactivity, low enantioselectivity, narrow substrate scope and competitive side reaction, i.e., double bond migration of the γ-hydroxyalkene encountered in palladium-catalyzed asymmetric alkene alkoxy-alkenylation of γ-hydroxyalkenes with alkenyl halides. We speculated that the suitable ligand and optimized reaction condition could serve as the key to solve these challenges. Herein we report our efforts on palladium-catalyzed asymmetric arylalkoxylation of γ-hydroxyalkenes with aryl halides to construct enantioenriched 2,2′-multisubstituted tetrahydrofuran derivatives (Scheme 1d).
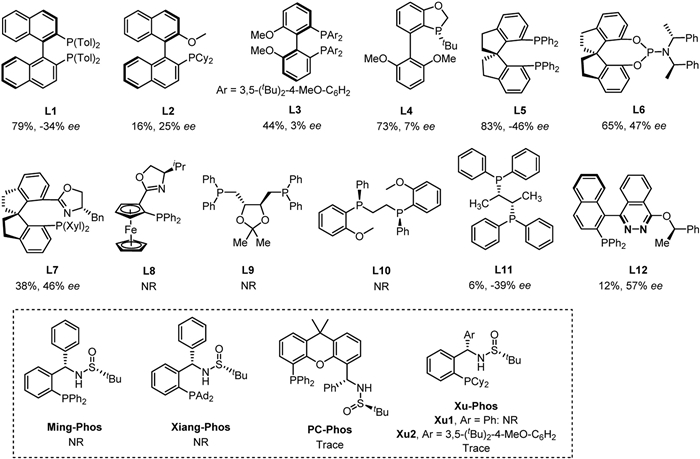
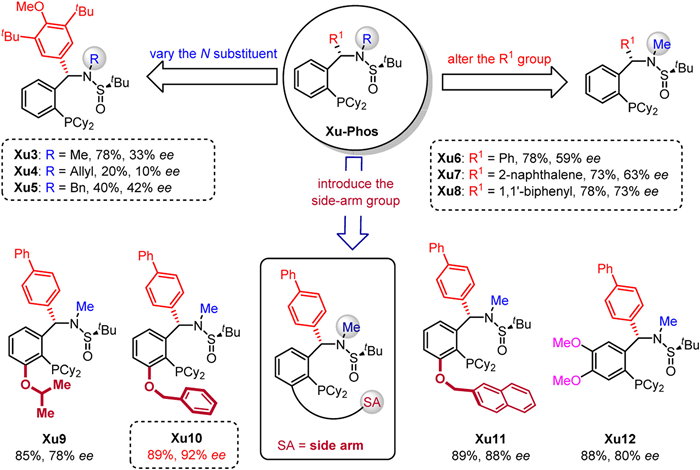
In our initial study, γ-hydroxyalkene 1a and aryl bromide 2a were selected as the model substrates. A series of privileged chiral ligands L1-L12 were investigated such as (R)-TolBINAP (L1), (R)-2-dicyclohexylphosphino-2′-methoxybiphenyl (L2), (R)-MeO-BIPHEP (L3), (R)-BIDIME (L4), spirochiral ligands (L5-L7), (R,R)-iPr-FOXAP (L8), (S,S)-DIOP (L9), (S,S)-Chiraphos (L11) and other chiral phosphine ligands (see more details in Supporting information). All these ligands failed to obtain the product 3a with good enantioselectivity except ligand L12 with 57% ee but in low yield (Fig. 1). Next, we turned attention to our developed sulfinamide-phosphine (Sadphos) ligands, such as Ming-Phos, Xiang-Phos, PC-Phos and Xu-Phos. Unfortunately, all of these ligands with free N—H bond were ineffective. To our delight, the N-Me-Xu-Phos (Xu3) could deliver desired product 3a in 78% yield albeit with 33% ee (Fig. 2), indicating that the variation of N-R group in Xu-Phos might tune the reactivity and enantioselectivity. With this hypothesis, we altered the N-R group to allyl (Xu4) and benzyl (Xu5) but both led to lower reactivity and only a little improvement of enantioselectivity was achieved with the Xu5 ligand. Further screening the N-Me-XuPhos ligand by adjusting the R1 group, the ee value substantially increased and the yield remained (e.g., Xu6: 78% yield, 59% ee; Xu7: 73% yield, 63% ee; Xu8: 78% yield, 73% ee). The ligand Xu9 with an iPrO side-arm could slightly improve the enantioselectivity from 73% ee to 78% ee compared with Xu8. Notably, the ligand Xu10 with a BnO side-arm could deliver much better enantioselectivity (92% ee) with higher reactivity (89% yield) [67]. However, when employing the bulkier naphthyl to supplant benzyl group, the ligand Xu11 gave relatively lower enantioselectivity. On the other hand, we also attempted to introduce electron-donating MeO groups at meta and para-positions of PCy2 group, which would enhance the electronic property of phosphine and facilitate oxidative addition of palladium to aryl bromides [68,69]. The ligand Xu12 improved the yield and the enantioselectivity slightly compared with the ligand Xu8. Subsequently, further comprehensive screening of the reaction conditions via changing additive, palladium salt, base, solvent, temperature, but all failed to further improve the enantioselectivity (Table 1).
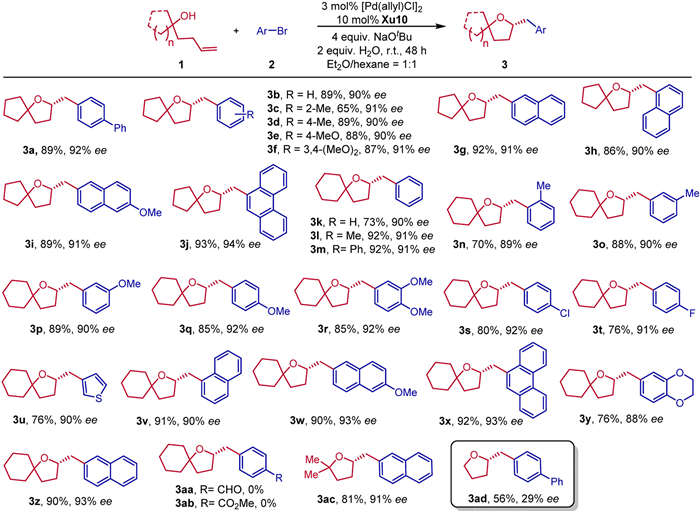
With the optimized reaction conditions in hand, we investigated the substrate scope of this transformation. A series of spirocyclic tetrahydrofurans were furnished in good yields with high enantioselectivities (Scheme 2). The substrate of 1-(but‑3-en-1-yl)-cyclopentan-1-ol 1a with various aryl bromides could work well to produce the desired products 3a-3j in 65%–93% yields with 90%–94% ee. Moreover, 1-(but‑3-en-1-yl)-cyclohexan-1-ol 1b with various aryl bromides, such as electron-donating (Me, MeO) groups at different positions of aryl bromides were well tolerated to afford the corresponding chiral spirocyclic tetrahydrofurans 3k-3q in 70%–89% yields with 89%–92% ee. Even 4-bromoveratrol furnished the corresponding product 3r in 85% yield with 92% ee. Generally, substrates with halogen atom (F or Cl) at the para-position of the aromatic ring reacted well to give the corresponding products 3s-3t in 76%–80% yields and 91%–92% ee. Modulation of the aryl skeleton of aryl bromides with a series of substituents, such as 3-thienyl, 1,4-benzodioxane-6-yl, phenanthren-9-yl, 2-naphthyl, and 1-naphthy, delivering the desired 3u-3z in 76%–92% yields with 88%–93% ee. It is a pity that no desired products 3aa and 3ab could be obtained by using aryl bromides bearing strong electron-withdrawing groups, such as aldehyde and ester groups. We proposed that the reduction elimination process is difficult to occur in the reaction of aryl electrophiles bearing electronically deficient groups. Fortunately, acyclic γ-hydroxyalkene 1c reacted smoothly with 2-bromonaphthalene to furnish the multisubstituted tetrahydrofuran 3ac in 81% yield with 91% ee. Notably, pent‑4-en-1-ol was also tolerated, giving 3ad in 56% yield with 29% ee. This result indicated that gem‑dialkyl groups at 2,2-positions in the hydroxyalkenes influenced strongly the stereoselectivity, which could be explained by the steric effect of gem‑dialkyl groups in favor of the formation of five-membered heterocycles.
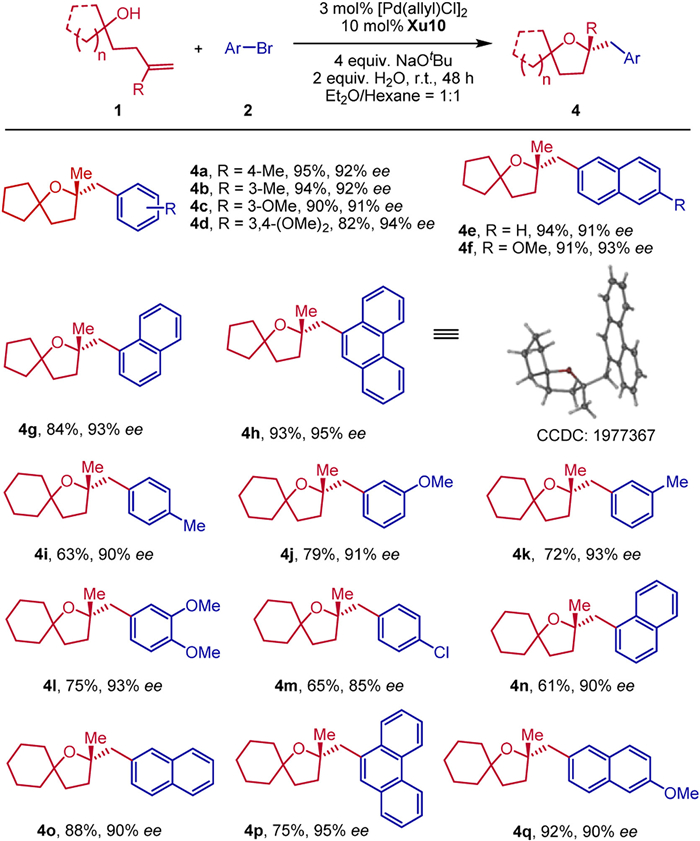
To further explore the scope of this reaction, a range of chiral spirocyclic tetrahydrofurans bearing a quaternary stereocenter were formed in excellent yields with high enantioselectivities (Scheme 3). For example, the substrate of 1-(3-methylbut-3-en-1-yl)-cyclopentan-1-ol 1d with various aryl bromides could work well to produce the desired products 4a-4h in 82%–95% yields with 90%–95% ee. Moreover, the reactions of 1-(3-methylbut-3-en-1-yl)cyclohexan-1-ol 1e with various aryl bromides bearing electron-donating groups (Me, OMe) at the meta- and para-position went well to furnish the corresponding products 4i-4l in 63%−79% yields with 90%–93% ee. The electron-withdrawing group such as halide (Cl) was compatible to afford the corresponding products 4m in 65% yield with 85% ee. 1-Naphthyl bromide, 2-naphthyl bromide and 9-bromophenathrene could also be employed to deliver the desired chiral spirocyclic tetrahydrofurans bearing a quaternary stereocenter 4n-4q in 61%–92% yields with 90%–95% ee.
Based on the studies by Wolfe and Tang as well as our previous work, the proposed catalytic cycle is shown in Scheme 4. The Pd0 species undergoes oxidative addition with the aryl bromide (2a) to afford PdⅡ complex Ⅱ. With NaOtBu as the base, ligand exchange between the PdⅡ complex Ⅱ and alcohol (1a) would lead to the formation of Pd–O bond and give intermediate Ⅲ. And intermediate Ⅲ undergoes migratory insertion, giving the Pd complex Ⅳ. This process was proposed as the stereoselectivity-determining step. Subsequent reductive elimination occurred to form the desired product 3a with concomitant regeneration of the Pd(0)L* catalyst. A stereochemical induction model is proposed. The BnO side-arm, the N-Me moiety, and the substituent R1 of Xu10 all contributed to this well-defined stereochemical process.

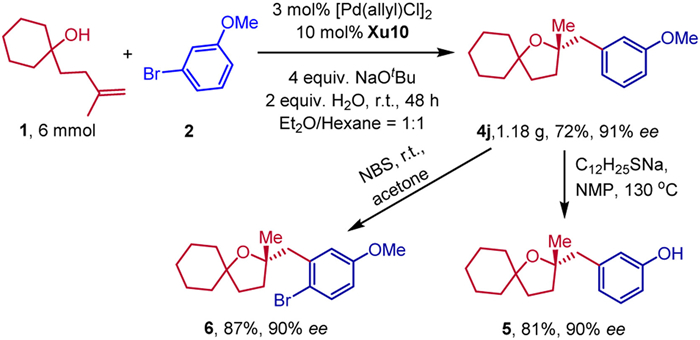
To display the synthetic utility of this methodology, a gram-scale reaction and derivatizations of 4j were accomplished (Scheme 5). Under standard conditions, 1.18 g of 4j was obtained in 72% yield with 91% ee. The enantiopure 4j could undergo a demethylation reaction to produce compound 5 with retention of the enantiomeric purity. Furthermore, a bromination product 4j was obtained in 87% yield with 90% ee.
In summary, we have developed an efficient palladium-catalyzed enantioselective arylalkoxylation of γ-hydroxyalkenes with aryl halides, which provided a facile access to a series of chiral tetrahydrofurans containing a tertiary or quaternary stereocenter in good yield with up to 95% ee. The salient features of this transformation include mild reaction conditions, readily available starting materials, a remarkable broad substrate scope, and good functional group tolerance. The chiral sulfnamidephosphine ligand Xu10 with a suitable side-arm was responsible for the high reactivity and enantioselectivity of this transformation. This study further confirmed that the introduction of a side-arm is an efficient strategy for tuning the enantioselectivity of a certain asymmetric reaction.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shuai Zhu: Data curation, Writing – original draft. Mingjie Chen: Data curation, Methodology. Haichao Shen: Data curation, Validation. Hanming Ding: Supervision. Wenbo Li: Supervision, Writing – review & editing. Junliang Zhang: Funding acquisition, Supervision, Writing – review & editing.
We gratefully acknowledge the funding support of the National Natural Science Foundation of China (NSFC, Nos. 22071060 and 22031004), the National Key R&D Program of China (No. 2021YFF0701600), and Shanghai Municipal Education Commission (No. 20212308).
Supplementary material associated with this article can be found, in the online version, at doi:
K.H. Jensen, M.S. Sigman, Org. Biomol. Chem. 6 (2008) 4083–4088. doi: 10.1039/b813246a
R.I. McDonald, G. Liu, S.S. Stahl, Chem. Rev. 111 (2011) 2981–3019. doi: 10.1021/cr100371y
H. Egami, M. Sodeoka, Angew. Chem. Int. Ed. 53 (2014) 8294–8308. doi: 10.1002/anie.201309260
E. Merino, C. Nevado, Chem. Soc. Rev. 43 (2014) 6598–6608. doi: 10.1039/C4CS00025K
G. Yin, X. Mu, G. Liu, Acc. Chem. Res. 49 (2016) 2413–2423. doi: 10.1021/acs.accounts.6b00328
S.W.M. Crossley, C. Obradors, R.M. Martinez, R.A. Shenvi, Chem. Rev. 116 (2016) 8912–9000. doi: 10.1021/acs.chemrev.6b00334
X.W. Lan, N.X. Wang, Y. Xing, Eur. J. Org. Chem. 2017 (2017) 5821–5851. doi: 10.1002/ejoc.201700678
R.K. Dhungana, S. KC, P. Basnet, R. Giri, Chem. Rec. 18 (2018) 1314–1340. doi: 10.1002/tcr.201700098
R. Giri, S. KC, J. Org. Chem. 83 (2018) 3013–3022. doi: 10.1021/acs.joc.7b03128
J.S. Zhang, L. Liu, T. Chen, L.B. Han, Chem. Asian J. 13 (2018) 2277–2291. doi: 10.1002/asia.201800647
J. Lin, R.J. Song, M. Hu, J.H. Li, Chem. Rec. 19 (2019) 440–451. doi: 10.1002/tcr.201800053
H. Jiang, A. Studer, Chem. Soc. Rev. 49 (2020) 1790–1811. doi: 10.1039/C9CS00692C
Y. Li, D. Wu, H. Cheng, G. Yin, Angew. Chem. Int. Ed. 59 (2020) 7990–8003. doi: 10.1002/anie.201913382
X. Fu, W. Zhao, Chin. J. Org. Chem. 39 (2019) 625–647. doi: 10.6023/cjoc201808031
C. Sun, G. Yin, Chin. Chem. Lett. 33 (2022) 5096–5100. doi: 10.1016/j.cclet.2022.04.026
Y. Wang, Y. Ping, W. Kong, Chin. Chem. Lett. 34 (2023) 108453–108458.
J.P. Wolfe, Eur. J. Org. Chem. 2007 (2007) 571–582. doi: 10.1002/ejoc.200600767
D.M. Schultz, J.P. Wolfe, Synthesis 44 (2012) 351–361. doi: 10.1055/s-0031-1289668
D.N. Mai, J.P. Wolfe, J. Am. Chem. Soc. 35 (2010) 12157–12159.
G.S. Lemen, J.P. Wolfe, Org. Lett. 12 (2010) 2322–2325. doi: 10.1021/ol1006828
E. Cahard, H.P.J. Male, M. Tissot, M.J. Gaunt, J. Am. Chem. Soc. 137 (2015) 7986–7989. doi: 10.1021/jacs.5b03937
M. Rigoulet, O. Thillaye du Boullay, A. Amgoune, D. Bourissou, Angew. Chem. Int. Ed. 59 (2020) 16625–16630. doi: 10.1002/anie.202006074
B. Peng, L. Dai, R. Liu, Org. Lett. 25 (2023) 2606–2610. doi: 10.1021/acs.orglett.3c00582
M.B. Hay, A.R. Hardin, J.P. Wolfe, J. Org. Chem. 70 (2005) 3099–3107.
L.T. Ball, M. Green, G.C. Lloyd-Jones, C.A. Russell, Org. Lett. 12 (2010) 4724–4727. doi: 10.1021/ol1019162
C. Zhu, J.R. Falck, Angew. Chem. Int. Ed. 50 (2011) 6626–6629. doi: 10.1002/anie.201101857
R. Zhu, S.L. Buchwald, J. Am. Chem. Soc. 137 (2015) 8069–8077. doi: 10.1021/jacs.5b04821
J. Gong, Q. Wang, J. Zhu, J. Am. Chem. Soc. 145 (2023) 15735–15741. doi: 10.1021/jacs.3c06158
W.B. Xie, Z. Li, Synthesis 52 (2020) 2127–2146. doi: 10.1055/s-0039-1690874
J.L. Kennemur, R. Maji, M.J. Scarf, B. List, Chem. Rev. 121 (2021) 14649–14681. doi: 10.1021/acs.chemrev.1c00620
H. Murayama, K. Nagao, H. Ohmiya, M. Sawamura, Org. Lett. 17 (2015) 2039–2041. doi: 10.1021/acs.orglett.5b00758
D. Chen, I.A. Berhane, S.R. Chemler, Org. Lett. 22 (2020) 7409–7414. doi: 10.1021/acs.orglett.0c01691
Y. Zhou, X. Xu, H. Sun, et al., Nat. Commun. 12 (2021) 1953–1963. doi: 10.1038/s41467-021-22287-w
I. Čorić, B. List, Nature 483 (2012) 315–319. doi: 10.1038/nature10932
Z. Sun, G.A. Winschel, A. Borovika, P. Nagorny, J. Am. Chem. Soc. 134 (2012) 8074–8077. doi: 10.1021/ja302704m
N. Tsuji, J.L. Kennemur, T. Buyck, et al., Science 359 (2018) 1501–1505. doi: 10.1126/science.aaq0445
J. Schlüter, M. Blazejak, F. Boeck, L. Hintermann, Angew. Chem. Int. Ed. 54 (2015) 4014–4017. doi: 10.1002/anie.201409252
P. Zhao, A. Cheng, X. Wang, et al., Chin J. Chem. 38 (2020) 565–569. doi: 10.1002/cjoc.201900544
Z. Yang, H. Li, S. Li, M.T. Zhang, S. Luo, Org. Chem. Front. 4 (2017) 1037–1041. doi: 10.1039/C6QO00806B
S.S. Gao, M. Garcia-Borràs, J.S. Barber, et al., J. Am. Chem. Soc. 139 (2017) 3639–3642. doi: 10.1021/jacs.7b01089
S.E. Denmark, W.E. Kuester, M.T. Burk, Angew. Chem. Int. Ed. 51 (2012) 10938–11095. doi: 10.1002/anie.201204347
C.Z. Yao, X.Q. Tu, H.J. Jiang, Q.K. Li, J. Yu, Tetrahedron Lett. 126 (2023) 154639–154650. doi: 10.1016/j.tetlet.2023.154639
S.H. Kang, S.B. Lee, C.M. Park, J. Am. Chem. Soc. 125 (2003) 15748–15749. doi: 10.1021/ja0369921
D.W. Tay, G.Y.C. Leung, Y.Y. Yeung, Angew. Chem. Int. Ed. 53 (2014) 5161–5164. doi: 10.1002/anie.201310136
U. Hennecke, C.H. Müller, C.G. Daniliuc, Eur. J. Org. Chem. 3 (2017) 484–490.
Y.F. Cheng, X.Y. Dong, Q.S. Gu, Z.L. Yu, X.Y. Liu, Angew. Chem. Int. Ed. 56 (2017) 8883–8886. doi: 10.1002/anie.201702925
Y.H. Jiang, D. Mondal, J.C. Lewis, ACS Catal. 12 (2022) 13501–13505. doi: 10.1021/acscatal.2c03383
Z.X. Zhan, X. Wang, J.T. Jiang, et al., Chin. Chem. Lett. 34 (2023) 107647–107651. doi: 10.1016/j.cclet.2022.06.070
L. Ma, X. Gao, X. Liu, et al., Chin. Chem. Lett. 34 (2023) 107735–107752.
M.T. Bovino, T.W. Liwosz, N.E. Kendel, et al., Angew. Chem. Int. Ed. 53 (2014) 6383–6387. doi: 10.1002/anie.201402462
Y. Miller, L. Miao, A.S. Hosseini, S.R. Chemler, J. Am. Chem. Soc. 134 (2012) 12149–12156. doi: 10.1021/ja3034075
J.P. Wolfe, M.A. Ross, J. Am. Chem. Soc. 126 (2004) 1620–1621. doi: 10.1021/ja0394838
B.A. Hopkins, Z.J. Garlets, J.P. Wolfe, Angew. Chem. Int. Ed. 54 (2015) 13390–13392. doi: 10.1002/anie.201506884
N. Hu, K. Li, Z. Wang, W. Tang, Angew. Chem. Int. Ed. 55 (2016) 5044–5048. doi: 10.1002/anie.201600379
S. Zhu, Z.H. Ye, M.J. Chen, et al., Nat. Commun. 14 (2023) 7611–7621. doi: 10.1038/s41467-023-43202-5
W. Zhou, X. Su, M.N. Tao, et al., Angew. Chem. Int. Ed. 54 (2015) 14853–14857. doi: 10.1002/anie.201508108
Z.M. Zhang, B. Xu, S. Xu, H.H. Wu, J.L. Zhang, Angew. Chem. Int. Ed. 55 (2016) 6324–6328. doi: 10.1002/anie.201602542
Z.M. Zhang, B. Xu, L.Z. Wu, et al., J. Am. Chem. Soc. 141 (2019) 8110–8115. doi: 10.1021/jacs.9b04332
Z.M. Zhang, B. Xu, Y.Y. Qian, et al., Angew. Chem. Int. Ed. 57 (2018) 10373–10377. doi: 10.1002/anie.201806372
Z.M. Zhang, P. Chen, W.B. Li, et al., Angew. Chem. Int. Ed. 53 (2014) 4350–4354. doi: 10.1002/anie.201401067
P.C. Zhang, Y.D. Wang, Z.M. Zhang, J.L. Zhang, Org. Lett. 20 (2018) 7049–7052. doi: 10.1021/acs.orglett.8b02999
Y.D. Wang, P.C. Zhang, X.Y. Di, et al., Angew. Chem. Int. Ed. 56 (2017) 15905–15909. doi: 10.1002/anie.201709595
L. Wang, M.J. Chen, P.C. Zhang, W.B. Li, J.L. Zhang, J. Am. Chem. Soc. 140 (2018) 3467–3473. doi: 10.1021/jacs.8b00178
H.X. Hu, Y.D. Wang, D. Qian, et al., Org. Chem. Front. 3 (2016) 759–763. doi: 10.1039/C6QO00087H
L. Wang, K.N. Zhang, Y.Z. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 4421–4427. doi: 10.1002/anie.201912408
H. Zhang, B. Xu, L.J. Zhou, Z.M. Zhang, J.L. Zhang, Green Synth. Catal. 5 (2024) 102–107. doi: 10.1016/j.gresc.2023.04.002
L.J. Wang, J. Zhou, Y. Tang, Chin. J. Chem. 36 (2018) 1123–1129. doi: 10.1002/cjoc.201800373
A. Aranyos, D.W. Old, A. Kiyomori, et al., J. Am. Chem. Soc. 121 (1999) 4369–4378. doi: 10.1021/ja990324r
C. Baillie, L.X. Zhang, J.L. Xiao, J. Org. Chem. 69 (2004) 7779–7782. doi: 10.1021/jo048963u

Scheme 1 Pd-catalyzed enantioselective alkene carboetherification reactions for synthesis of chiral O-heterocycles.

Scheme 2 Scope with respect to the aryl halides. Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), NaOtBu (4 equiv.), H2O (2 equiv.), 3 mol% [Pd(allyl)Cl]2, and 10 mol% ligand in 2.0 mL Et2O/hexane = 1:1, room temperature, under N2 for 48 h. The yield is isolated yield. ee was determined by chiral HPLC analysis.

Scheme 3 Enantioselective formation of quaternary centers. Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), NaOtBu (4 equiv.), H2O (2 equiv.), 3 mol% [Pd(allyl)Cl]2, and 10 mol% ligand in 2.0 mL Et2O/hexane = 1:1, room temperature, under N2 for 48 h. The yield is isolated yield. ee was determined by chiral HPLC analysis.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



