Key Laboratory of Optic-electric Sensing and Analytical Chemistry for Life Science, MOE, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
b.
Laboratory of Catalytic Conversion and Clean Energy in Universities of Shandong Province, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, China
c.
Department of Medicinal Chemistry, School of Pharmacy, Hengyang Medical School, University of South China, Hengyang 421001, China
Received Date:
26 December 2023 Accepted Date:
19 February 2024 Revised Date:
01 February 2024 Available Online:
15 November 2024
Abstract:
Developing applicable methods to forge linkages between sp3 and sp2-hydridized carbons is of great significance in drug discovery. We show here a new, Ni-catalyzed reductive cross-coupling reaction that forms Csp3−Csp2 bonds from aryl iodides and cyclic sulfonium salts. Notably, Csp3−Csp2 bonds can be forged selectively at the iodine-bearing carbon of bromo(iodo)arenes which is usually recognized as a huge challenge under the catalytic reductive cross-coupling (CRCC) conditions. Experimental and computational mechanistic studies support LNiⅠAr as an active species, while the untraditional anti-Markovnikov selective alkylation of asymmetric sulfonium salts is determined by the oxidative S-substitution of sulfonium salts with LNiⅠAr. This protocol further expands the range of alkyl electrophiles under the CRCC conditions and provides a new strategy for the construction of Csp3−Csp2 bonds.
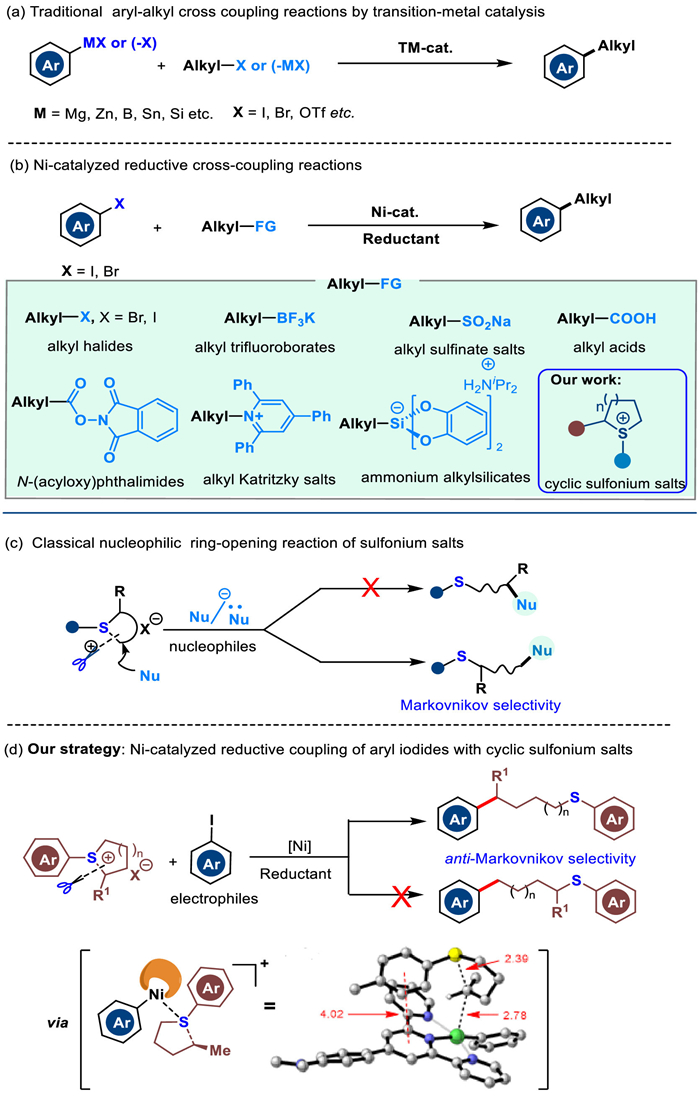
The degree of saturation of organic molecules has a great influence on their physical properties, such as crystallinity and aqueous solubility [1–4]. Recent research has shown that an increased fraction of sp3-hydridized carbons, and thus reducing its planarity, is an effective approach to make the compound more drug-like [5–7]. Consequently, there is a continued interest in developing concise and widely applicable methods to forge linkages between sp3 and sp2-hydridized carbons in drug discovery [8,9]. Traditionally, transition metal-catalyzed cross-coupling reactions between arylmetallic species and alkyl halides, and transition metal-catalyzed cross-coupling of aryl halides with alkylmetallic reagents, are the two classical methods for constructing Csp3−Csp2 bonds (Scheme 1a) [10–19]. Over the past decade, the nickel-catalyzed reductive cross-coupling reactions have emerged as a powerful strategy to forge Csp3−Csp2 bonds in the presence of a terminal reductant. In this field, the pioneering work has been reported by Périchon [20], Knochel [21], Gosmini [22], Lipschutz [23], Jacobi von Wangelin [24], Weix, [8,25–28] MacMillan, [29,30] Buchwald, [31] Molander [32,33], Gong [34,35], and others [36–41]. Notably, in this respect, the alkylation reagents mainly focus on alkyl halides [42–45], alkyl trifluoroborates [46,47], ammonium alkylsilicates [48,49], alkyl Katritzky salts [33], N-(acyloxy)phthalimides [50], alkyl acids [51,52], and alkyl sulfinate salts [53], and so on [54,55] (Scheme 1b). Although great achievements have been made in this field, the stoichiometric organometallic reagents, harsh reaction conditions, and unavailability of starting materials restricted their wide applications in organic synthesis. Therefore, it is still highly desirable to develop more robust methods to access Csp3−Csp2 bonds directly from stable and readily available starting materials under catalytic reductive cross-coupling (CRCC) conditions.
Scheme 1
Scheme 1.
(a) Traditional aryl-alkyl cross-coupling reactions by transition-metal catalysis. (b) Ni-catalyzed reductive cross-coupling reactions. (c) Classical nucleophilic ring-opening reaction of sulfonium salts. (d) Invention of a new Ni-catalyzed reductive coupling of aryl iodides with cyclic sulfonium salts.
Sulfonium salts are an important class of sulfur-containing reagents, which have long been used as precursors to sulfur ylides and play a vital role in the construction of cyclic compounds as one-carbon building blocks [56]. On the other hand, the nucleophilic Markovnikov ring opening reactions of sulfonium salts with nucleophiles have also been sporadically reported (Scheme 1c) [57,58]. In recent years using sulfonium salts as radical precursors for constructing C−C bonds and C−heteroatom bonds has been well developed [59–72]. However, the reductive coupling reactions using sulfonium salts as electrophiles are rarely reported. In 2021, Ritter and co-workers initially reported an elegant palladium-catalyzed reductive electrophile cross-coupling of aryl thianthrenium salts with alkyl iodides for the construction of Csp3−Csp2 bonds under mild conditions [73,74]. However, developing novel reductive coupling modes of sulfonium salts as electrophiles still remains a huge challenge.
The aryl alkyl thioether skeletons are ubiquitous in natural products, pharmaceuticals, and biologically active molecules [75–77]. Therefore, the development of new reaction modes of sulfonium salts and the introduction of alkylthio fragments into organic molecules is of great significance in the field of drug development. With the insight obtained from our recent experimental results in the radical type ring-opening reactions with sulfonium salts [78,79], we envision that Csp3−Csp2 bonds could be formed starting from readily available aryl iodides and cyclic alkyl(aryl)sulfonium salts under CRCC conditions. We speculate that, during the C−S bond cleavage of alkyl sulfonium salts, the combination of bulky tridentate ligand and radical-containing nickel complexes possibly obstacles the coordination of alkyl moiety to nickel center and affords an alkyl radical species. In this scenario, different to the nucleophilic ring opening, anti-Markovnikov selectivity might be realized (Scheme 1d). To the best of our knowledge, the anti-Markovnikov selective ring-opening reaction of cyclic alkyl(aryl)sulfonium salts under CRCC conditions has never been documented to date, and how to avoid the Csp2−S bond cleavage under Ni catalysis will be a huge challenge for our protocol [59].
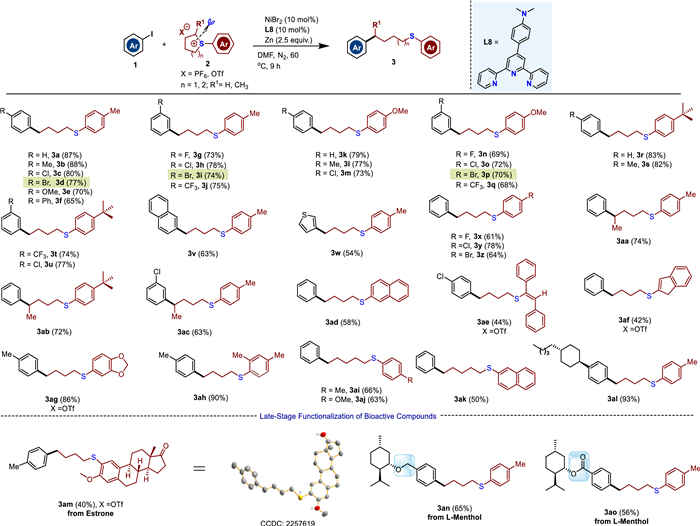
To validate our hypothesis, our investigation of this CRCC transformation was performed between iodobenzene 1a and sulfonium salt 2a. As shown in Table 1, five nickel catalysts such as NiBr2, NiCl2(dppe), NiCl2, NiI2, and Ni(OAc)2·H2O were examined in DMF in the presence of 2.5 equiv. of Zn powder at 60 ℃ (entries 1−5), and NiBr2 provided the highest yield 75% (entry 1). Next, various ligands including tridentate ligands (L1−L9) and bidentate ligands (L10−L14) were investigated, and 4′-(p-dimethylaminophenyl)−2,2′:6′,2′′-terpyridine L8 showed the highest activity, affording the desired product 3a in 87% yield with the corresponding homocoupling product 3b' in a trace amount (entries 6 and 7, see Supporting information for details). Then, we tested various solvents including DMF, NMP, DMSO, toluene, THF, and DMF was superior to the others (compare entries 6, 8−11). Furthermore, other reductants, including Mn, Mg, DEMS and TES were examined and resulted in lower productivity than Zn (entries 12–16). Finally, controlled experiments show that reducing the temperature can lead to a decrease in the yield of the target product, and accelerate the temperature cannot improve the yield (entrie 16–18).
With the optimized reaction conditions in hand, we next began to investigate the scope and generality of the nickel-catalyzed reductive alkylation of aryl iodides with cyclic alkyl(aryl)sulfonium salts, and the results are listed in Scheme 2. We were pleased to find that diverse aryl iodides 1 containing either electron-rich or electron-poor groups smoothly reacted with sulfonium salts 2 affording the corresponding sulfur-containing alkylation products in moderate to good yields. The structure of the alkylation products was determined by the single crystal X-ray diffraction study of compound 3am. It is worth mentioning that a range of bromo(iodo)arenes bearing iodo and bromo groups in the meta- and para-positions reacted in good yields to afford the desired products with excellent chemoselectivity (3d, 3i and 3p). A heterocycle iodide such as 3-iodothiophene delivered the product 3w in 54% yield. Of note that this reductive cross-coupling transformation has a different regioselectivity from the nucleophilic ring-opening reaction, the alkylation products were obtained in an anti-Markovnikov pathway at a higher steric hindrance site (3aa−3ac). Experimental and computational mechanistic studies support LNiⅠAr as an active species, while the untraditional anti-Markovnikov selective alkylation of asymmetric sulfonium salts is determined by the oxidative S-substitution of sulfonium salts with LNiⅠAr (Fig. 1). Further exploration on the scopes and limits of the synthetic application are in progress. To our delight, the styrene-substituted sulfonium salt was also suitable for the reaction affording 3ae in 44% yield. In addition, six-membered cyclosulfonium salts could also efficiently participate in the catalytic system delivering the desired products in good yields (3ai−3ak). Biologically relevant molecules such as estrone and L-menthol were well functionalized with moderate yields (3am−3ao) which demonstrated the practical application of the reaction. Unfortunately, we found that treatment of iodobenzene 1a with thianthrenium salts or acyclic aryl sulfonium salts under the standard reaction conditions delivered no desired products (see Supporting information for details). Further exploration on the scopes and limits of the synthetic application are in progress. A lot of functional groups were tolerated in the present transformation, such as halogen substituents, methyl, and methoxy, leaving ample room for further modifications.
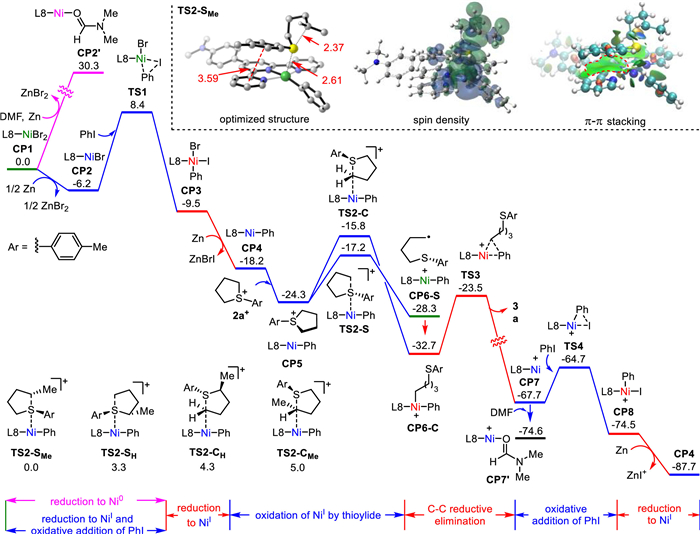
Figure 1.
Calculated solution-phase Gibbs free energy profile of Ni-catalyzed cross-coupling of PhI and sulfonium salt 2a (in kcal/mol). Optimized structure of TS2-SMe was provided with most of hydrogen atoms omitted for clarity. Bond distances and the minimum distance between the geometric centers of the benzene ring on S atom and the pyridine rings of ligands were given in angstrom (red). Mulliken spin density and plot of noncovalent interaction were made at the isosurface value of 0.0004 and 0.03, respectively. Pink, blue, green and red were used to denote Ni0, NiⅠ, NiⅡ and NiⅢ, respectively.
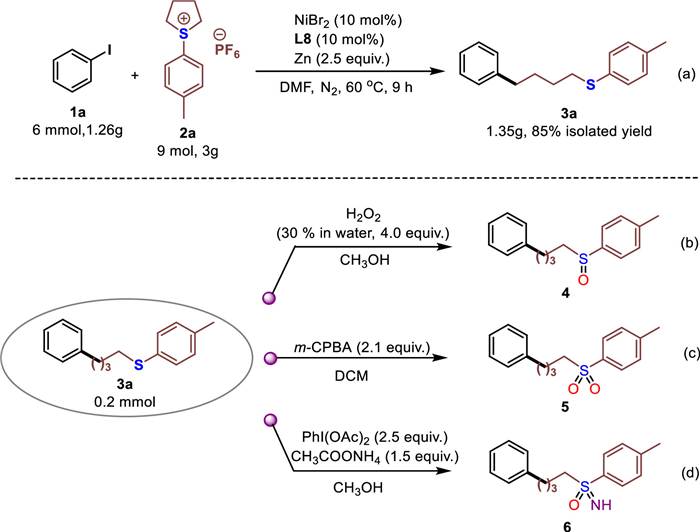
We explored the synthetic utility of this reductive cross-coupling protocol. The gram-scale reaction was performed between 1a and 2a, and the reaction gave 3a in 85% isolated yield without loss of reactive efficiency (Scheme 3a). In addition, the sulfoxide product 4, sulfone product 5 and analogues sulfoximine product 6 were efficiently synthesized under different oxidative conditions (Schemes 3b-d). Thus, this simple Ni-catalyzed reductive cross-coupling protocol could be served as an alternative method for the synthesis of sulfur-containing organic derivatives.
To gain insight into the mechanism of this reaction, several control experiments were conducted. Firstly, the reaction did not proceed in the absence of Zn (Scheme 4a). Furthermore, when the reaction was performed using Ni(cod)2 as the catalyst, no alkylation product 3b was detected, but the alkyl dimer 3b' was obtained as the major product (Scheme 4b). These preliminary results suggest that the transformation is not initiated by nickel(Ⅱ) or nickel(0) catalyst, it could occur with Ni(Ⅰ) species, which is in accordance with the previous reports [20]. To further elucidate the reaction pathway, a tridentate Ni(Ⅱ) complex 7 was obtained by reaction of Ni(cod)2 with methyl 4-iodotoluene in the presence of L8. No desired 3b was detected for the reactions of 1b with 2a using 7 as the precatalyst, in the absence of Zn. While, with Zn, 3b was obtained in 85% yield (Scheme 4c). We reason that it is likely that the complex 7 was reduced by Zn to L8Ni(Ⅰ)-Ar, which reacts with 2a to give Ar-Ni(Ⅲ)alkyl prior to the reductive elimination delivering 3b.
Scheme 4
Scheme 4.
Investigations of the reaction mechanism.
Furthermore, the mechanistic details were investigated with the aid of density functional theory (DFT) calculations at the level of B3LYP-D3/SMD (solvent = DMF)/SDD&def2-TZVP//B3LYP-D3/SMD (solvent = DMF)/LANL2DZ+p&6–31G(d) (see Supporting information for more computational details). As shown in Fig. 1, reduction of NiBr2 complex CP1 to Ni0 complex CP2′ with Zn is highly endergonic by 34.8 kcal/mol, being significantly less favored than the reduction to Ni(Ⅰ) complex CP2. This result is consistent with our control experiment which excluded a Ni0-NiⅡ catalytic process (Scheme 4b). Very recently, the experimental mechanistic study from Li also indicates that the reduction of NiI2 precatalyst by Zn generates NiⅠ species rather than Ni0 species [20]. From CP2, oxidative addition of PhI goes through TS1 to afford NiⅢ complex CP3 which is then again reduced to NiⅠ−pH complex CP4 by Zn. Thereafter, sulfonium cation 2a+ can oxidize the NiⅠ species via C-substitution transition state TS2-C or S-substitution transition state TS2-S. For 2a+, TS2-S is slightly favored over TS2-C by 1.4 kcal/mol and generates a NiⅡ complex CP6-S that contains an alkyl radical (Fig. S1 in Supporting information for the plot of Mulliken spin density). The bonding of the alkyl radical to NiⅡ center forms the more stable NiⅢ complex CP6-C, from which C(sp3)−C(sp2) reductive elimination can occur viaTS3. The resulting NiⅠ complex CP7 undergoes oxidative addition with another PhI viaTS4, followed by reduction with Zn to CP4 to restart the catalytic cycle. According to the calculated energy profile, the oxidation of NiⅠ−pH complex CP4 determines the regioselectivity for asymmetric sulfonium salts (Scheme 2, 3aa−3ac). Taking the starting material of product 3aa as the model substrate, the S-substitution transition states (TS2-SMe and TS2-SH) also have lower energies than the C-substitution transition states (TS2-CMe and TS2-CH). This phenomenon possibly results from the fact that the benzene ring on the S atom is closer to the pyridine rings of ligand L8 in TS2-SMe and TS2-SH as reflected by the minimum distance of their geometric centers (Fig. 1 and Fig. S1). Thus a stronger π-π stacking effect is expected in TS2-SMe and TS2-SH (Fig. 1 and Fig. S1 for plots of noncovalent interaction). On the other hand, TS2-SMe is more stable than TS2-SH by 3.3 kcal/mol, supporting the superiority of C−S bond cleavage at the alkyl-substituted C-terminal. It was found that remarkable Mulliken spin density spreads on the cleaving carbon in TS2-SMe and TS2-SH with the absolute values of 0.495 and 0.561, respectively, contributing to the lower energy of TS2-SMe that owns a more stable carbon radical.
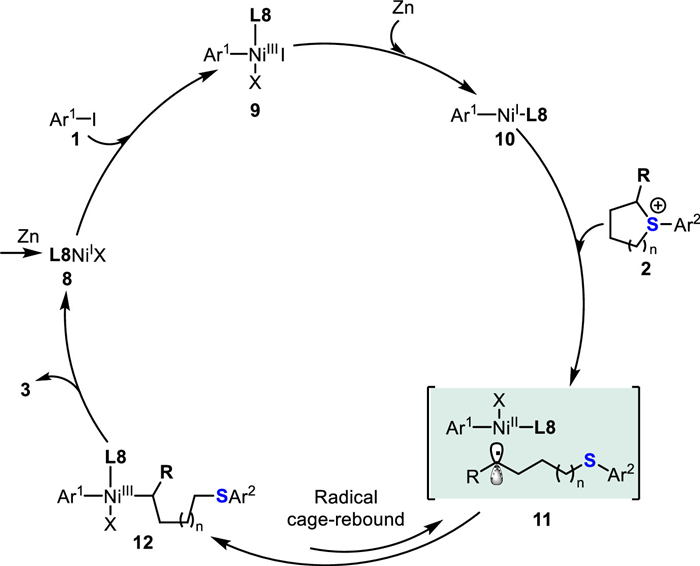
According to the mechanistic studies, a proposed catalytic cycle is proposed in Scheme 5. Firstly, the reduction of L8NiⅡ precatalyst by Zn generates NiⅠ species 8. Then, oxidative addition of aryl iodides 1 to NiⅠ gives a NiⅢ species 9 that is subjected to reduction by stoichiometric Zn leading to Ar1−NiⅠ−L8 complex 10. Subsequently, the complex 10 acts as a reducing agent and delivers an electron to 2, thus giving a formal Ar1−NiⅢ−C(sp3) species 12 that undergoes an in-cage radical/Ni rebound 11 [80]. Finally, reductive elimination of the species 11 affords the desired cross-coupling product 3 while regenerating the NiⅠ catalyst to restart the next catalytic cycle.
In conclusion, we have successfully developed an efficient method for Csp3−Csp2 bonds formation based on a Ni-catalyzed reductive cross-coupling protocol using cyclic sulfonium salts as alkylation reagents. This reaction tolerates a broad scope of functional groups and provides the corresponding aryl alkyl thioethers in moderate to good yields. This method further expands the range of alkyl electrophiles under the CRCC conditions and provides a new strategy for the construction of Csp3−Csp2 bonds. This protocol also provides a new avenue to aryl alkyl thioethers using cyclic sulfonium salts as alkylthio fragments surrogates. Importantly, the utility of this method was proven by late-stage functionalization of bioactive molecules.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 22271170), and the Scientific Research Foundation of Qingdao University of Science and Technology.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109732.
[1]
A.W. Dombrowski, N.J. Gesmundo, A.L. Aguirre, et al., ACS Med. Chem. Lett. 11 (2020) 597–604. doi: 10.1021/acsmedchemlett.0c00093
[2]
H.Y. Song, J. Jiang, Y.H. Song, et al., Chin. Chem. Lett. 35 (2023) 109246.
J. Zhou, D. Wang, W. Xu, et al., J. Am. Chem. Soc. 145 (2023) 2081–2087. doi: 10.1021/jacs.2c13220
Scheme 1
(a) Traditional aryl-alkyl cross-coupling reactions by transition-metal catalysis. (b) Ni-catalyzed reductive cross-coupling reactions. (c) Classical nucleophilic ring-opening reaction of sulfonium salts. (d) Invention of a new Ni-catalyzed reductive coupling of aryl iodides with cyclic sulfonium salts.
Figure 1
Calculated solution-phase Gibbs free energy profile of Ni-catalyzed cross-coupling of PhI and sulfonium salt 2a (in kcal/mol). Optimized structure of TS2-SMe was provided with most of hydrogen atoms omitted for clarity. Bond distances and the minimum distance between the geometric centers of the benzene ring on S atom and the pyridine rings of ligands were given in angstrom (red). Mulliken spin density and plot of noncovalent interaction were made at the isosurface value of 0.0004 and 0.03, respectively. Pink, blue, green and red were used to denote Ni0, NiⅠ, NiⅡ and NiⅢ, respectively.

 DownLoad:
DownLoad:



 下载:
下载:







