School of Chemistry and Chemical Engineering, Zhejiang Sci-Tech University, Hangzhou 310000, China
* Corresponding author. E-mail address: shenchengshuo@zstu.edu.cn (C. Shen). 1 These authors contributed equally to this work.
Received Date:
13 December 2023 Accepted Date:
22 February 2024 Revised Date:
04 February 2024 Available Online:
15 November 2024
Abstract:
Nanographenes (NGs) with twisted backbones are emerging as new candidates for chiroptical materials. In this work, we describe a new strategy for synthesizing a [10]twistacene-embedded NG which exhibits a rare flag-hinge-like geometry. By neatly creating steric crowding on the [6]helicene breaches of the NG skeleton, the synthesis only provided homochiral isomers without generating the "meso‑" isomer. The formed NGs showed high luminescence with quantum yield up to 52%, and promising circularly polarized luminescence (CPL) performance with |glum| up to 5.0 × 10−3. Besides, these NGs also showed outstanding CPL brightness (BCPL) up to 305 L mol−1 cm−1 among chiral NGs.
Circularly polarized luminescence (CPL) materials have captivated considerable attention in the last decades owing to the potential applications in the fields of optoelectronic devices [1–3], telecommunication [4,5], security [6,7], 3D imaging [8], as well as smart sensors/probers [9–11]. Among all CPL materials, chiral small organic molecules have been widely investigated due to the accessibility to straightforward and rational designing and tuning of their fluorescence intensities, colors and CPL dissymmetry factors [12–14]. Most CPL organic molecules could be categorized into several sorts including chiral ketones, helicenes, biaryls, paracyclophanes as well as BODIPYs [15–19]. Meanwhile, chiral nanographenes (NGs) have emerged as novel CPL materials recently for their excellent luminescence properties [20–27]. For most chiral NGs, helicene units were used embedded in the π-conjugated backbones, making NGs with unique helical geometries. For instance, superhelicenes with lateral π-extension from a helicene core have been largely reported, showing a 2D-extended helicoid topology [26,28–31]. Besides, orderly circling two or more helicene units in the NG skeletons gave a rich array of propeller-shaped chiral NGs [20,32–39]. In several cases, multiple helicene units could also endow chiral NGs with layered structures [40–42]. On the other hand, examples by employing asymmetric sp3 carbons or rotation-blocked C—C bonds have demonstrated alternative portals to chiral NGs with central or axial chirality [43–45]. These chiral geometries have enriched the skeleton of chiral NGs, thus providing easy access to studying the relationship between the chiral geometries and their chiroptical properties.
Recently, twisted chiral NGs have been demonstrated with distinct out-of-plane deformation geometry and intriguing chiroptical properties [46]. Among these NGs, twistacenes have been pioneeringly investigated [47]. Their inherent chirality could be generated from over-crowding of excessive phenyl rings in the periphery [48–50], or from neatly designed mechanical locking [51]. Besides, a few examples have also demonstrated that the twist geometry could be engendered from non-sextet cycles [24,52]. Owing to the easy synthetic access, introducing multiple helicene breaches in the ribbon-like NGs showed a more favorable way to synthesize larger twisted chiral NGs [53–56]. For example, by employing eight [4]helicene moieties in both lateral sides of ribbon-like NGs, [10]twistacene-embedded NG with an overall twist angle of 170° has been reported [54]. Similarly, by introducing [5]helicene units, a series of super twistacenes were prepared with outstanding chiroptical properties [55,56]. However, this strategy also brought new challenges that how to keep these helicenes with identical handedness, thus promoting the twisting of the NG backbones (with all the helicenes in P- or M- handedness), without forming other unappealing diastereoisomers such as the "meso‑" form. For NGs with [4]helicene breaches, introducing chiral auxiliary could tune the flexible [4]helicenes with the same handedness [57,58]. While for twisted chiral NGs with [5]helicene breaches or even larger ones, synthetic control revealed to be difficult, and in several cases, the meso‑form revealed to be the major product [53]. Steric control in the synthesis revealed to be an efficient way to generate homochiral isomers in synthesis of several propeller-shaped chiral NGs [37,59], and herein we adopted this strategy in the synthesis of twisted NGs and synthesized a [10]twistacene-embedded chiral NG containing two [6]helicene breaches (NG1) which was generated in a homochiral fashion. Theoretical calculation indicated that the bulkiness of the tert‑butyl groups on the [6]helicene breaches assisted the formation of the homochiral NG1 rather than the heterochiral one. The formed twisted NG showed a large end-to-end twist angle of ca. 72°, making the whole molecule in a rare flag-hinge-like pattern. Besides, NG1, together with its shorter homologue NG2, showed prominent luminescence properties with high quantum yield up to 52%, and high CPL dissymmetry factor |glum| up to 5.0 × 10−3. The enantiopure NG1 also showed an outstanding CPL brightness BCPL up to 305 L mol−1 cm−1, which is considerably high among the reported NGs [60]. This work provides a new concept of designing highly luminescent chiral NGs, and might stimulate their prospective chiroptical applications in the future.
The preparation of NG1 is briefly depicted in Fig. 1a. The synthesis was started from Suzuki cross-coupling between a dibromophenyl substituted pyrene 1 and two hexabenzocorannulene (HBC) boronic ester 2, giving the uncyclized precursor 3 with a yield of 45%. Subsequently, Scholl cyclization of 3 in the presence of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and CF3SO3H gave a dark red powder NG1 in a plausible yield of 94%. The cyclization of two C—C bonds with a total loss of four hydrogen atoms from precursor 3 to NG1 could be proved by the mass spectroscopy (Figs. S11 and S13 in Supporting information). For the obtained NG1, we at first considered that the reaction might form two diastereoisomers (homochiral and heterochiral ones) since the molecule contains two [6]helicene units. However, simple purification by short silica column chromatography gave the product with only one set of peaks in a high symmetry according to the 1H NMR analysis. This result indicated that the Scholl cyclization might merely generate one of the diastereoisomers. To further unveil the exact structure of the obtained NG1, we tried to grow single crystals through slow diffusion of methanol vapor into the NG1 solution in THF, and dark-red tabular single crystals were obtained. X-ray diffraction showed that NG1 adopted a homochiral geometry, and was crystalized in a racemic form deposited in a P1 space group (Fig. 1b). Each NG1 molecule displayed a homochiral structure with both [6]helicene breaches in a (P,P)- or (M,M)-form. Notably, NG1 contained a twisted [10]acene moiety with a length of 24.6 Å. Besides, the two [6]helicene breaches divided the nanographene into three parts – two HBC units and one peropyrene unit. In the side-view, we found a large end-to-end twist angle of 72°, providing the whole skeleton in a flag-hinge-like geometry (Fig. 1d). This might be probably owing to the large interplanar angles of the two terminal rings of [6]helicene that stretched the skeleton [61]. Besides, tert‑butyl groups on the [6]helicene moieties were found covering the two sides of the central peropyrene unit, forming obvious van der Waals interactions according to the Independent Gradient Model based on Hirshfeld partition of molecular density (IGMH) analysis (Fig. 1c, green area) [62], and owing to the shielding effect, these two tert‑butyl groups exhibited more shielded signals (−0.03 ppm) than other tert‑butyl groups (1.79 ppm to 2.05 ppm) in the 1H NMR experiment (Fig. S8 in Supporting information). In the crystal packings, (P,P)- and (M,M)-NG1 assembled alternatively, and owing to the abundant tert‑butyl groups, the crystal majorly packed via van der Waals contacts, and only weak intermolecular π-π interaction was found between two HBC units with the distance of 3.7 Å (Fig. 1d). Furthermore, we also prepared the smaller homologue nanographene NG2 with only one side fused with the HBC unit (Fig. 1a).
Figure 1
Figure 1.
(a) Synthetic routes for NG1 and NG2. (b) Single-crystal structure of NG1 with thermal ellopsoids shown at 30% probability (Hydrogen atoms are omitted for clarity). [10]twistacene unit is shown in red. (c) IGMH analysis of NG1 between tert‑butyl groups and central perylene core (Geometry was optimized at the PBE0-D3(BJ)/def2-SVP level of theory). (d) Side-view of single-crystal structure of NG1 with tert‑butyl groups and hydrogen atoms omitted, and packings of (P,P)-NG1 (in red) and (M,M)-NG1 (in green) in the unit cell.
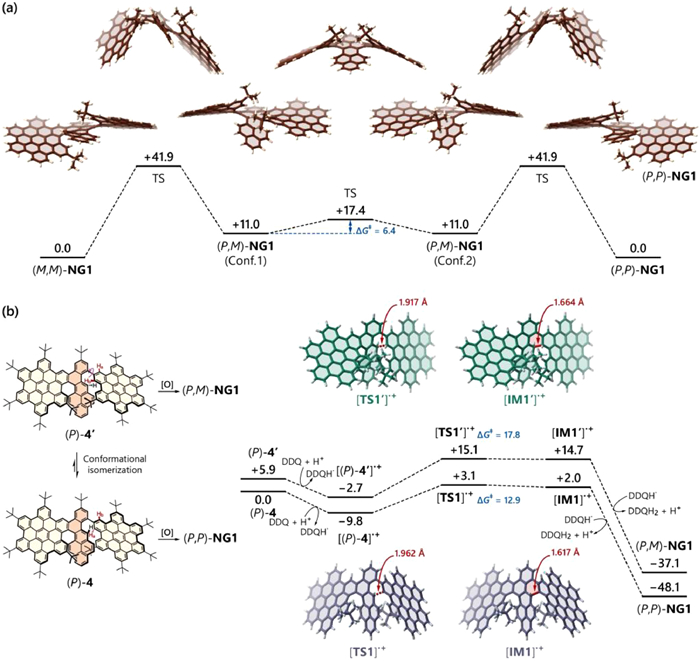
To further understand the high synthetic selectivity only generating the homochiral isomer rather than the heterochiral one in the case of NG1, we investigated its intrinsic mechanism using DFT calculations at the PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)-def2-SVP level of theory (nine less relevant peripheral tert‑butyl groups were replaced with hydrogen atoms for calculation simplicity). Firstly, we optimized the local minima of both homochiral (P,P)-NG1 and heterochiral (P,M)-NG1, and the corresponding transition states involved in the isomerization process (Fig. 2a). Notably, the heterochiral one was not exactly a meso‑form owing to the steric repulsion of two adjacent tert‑butyl groups. The two mirror-image conformers showed a low conversion barrier ΔG‡ of 6.4 kcal/mol.
Figure 2
Figure 2.
(a) Isomerization process between homochiral and heterochiral NG1 isomers. (b) Cyclization process from 4 to NG1 isomers. The calculations were performed in the PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP level, and the Gibbs free energies are given in kcal/mol. Peripheral tert‑butyl groups were omitted for calculation simplicity.
The free energy of heterochiral (P,M)-NG1 revealed 11.0 kcal/mol higher than the homochiral (P,P)-NG1, suggesting that the homochiral isomers are much more stable than heterochiral ones thermodynamically. This was probably attributed from the steric repulsion of the two tert‑butyl groups in the case of (P,M)- NG1 (Calculation on the isomers of NG1 without any tert‑butyl groups gave a free energy difference of only 0.6 kcal/mol (Fig. S29 in Supporting information). The isomerization barrier ΔG‡ from (P,P)- to (P,M)-NG1 was calculated as 41.9 kcal/mol, which indicated a high configurational stability of NG1 against diastereoisomerization or racemization. Subsequently, we delved into the detailed reaction process of the Scholl reaction on the final cyclization (Fig. 2b). The calculation started from mono-cyclized intermediate (P)−4 where the two inner tert‑butyl groups were at the opposite sides. Due to the free rotation of one uncyclized HBC unit, (P)−4 could convert to another conformer (P)−4′ where the two inner tert‑butyl groups were situated at the same side. (P)−4′ showed a free energy of 5.9 kcal/mol higher than the original conformer (P)−4. The reaction process was calculated employing the radical cation mechanism [63,64]. For the first step, the starting (P)−4 lost one electron, forming the radical cation [(P)−4]·+. Then, the uncyclized [(P)−4]·+ was converted into the cyclized [IM1]·+via a reaction barrier of 12.9 kcal/mol. After losing of two hydrogen atoms and one additional electron, the cyclized [IM1]·+ was finally transformed to the product (P,P)-NG1 with total free energy release of 48.1 kcal/mol. Meanwhile, in the pathway from conformer (P)−4′ to (P,M)-NG1, the cyclization process showed a higher reaction barrier of 17.8 kcal/mol compared with (P)−4 pathway. Besides, the transition state TS1' also possessed a higher free energy of 12.0 kcal/mol, relatively. This result indicated that the Scholl reaction for the second cyclization might selectively form the homochiral isomers (P,P- or M,M-) rather than the heterochiral one (P,M-), thus echoing the experimental result that only the homochiral NG1 has been obtained. In case of NG2, the racemization barrier was calculated as 37.5 kcal/mol. Owing to the high isomerization barrier of NG1 and NG2, we were able to resolve both enantiomers via chiral high-performance liquid chromatography (HPLC) by employing CHIRAPAK IB column or CHIRAPAK IF column with ee value higher than 98% (Figs. S15 and S18 in Supporting information).
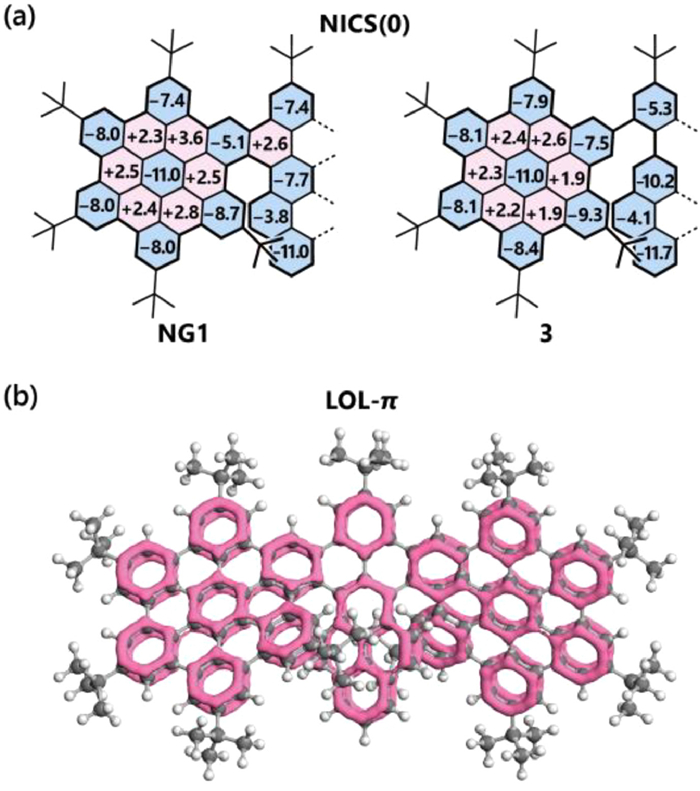
The aromaticity of both NG1 and 3 was investigated via nucleus-independent chemical shift (NICS) [65,66] and localized orbital locator (LOL) [67,68] analyses (Fig. 3). For the NICS analysis, we found NG1 generally exhibits similar aromaticity as its precursor 3, but with values largely changed for the rings close to the newly formed benzenoid cycle. For example, the adjacent three sextets showed NICS(0) values of −5.3, −10.2 and −7.5 in 3, while it changed to −7.4, −7.7 and −5.1, respectively after the cyclization. Besides, the LOL-π analysis provided similar results that the π-cycles are localized on the rings with negative NICS(0) values, indicating these rings with higher aromaticity.
Figure 3
Figure 3.
(a) NICS(0) values of NG1 and precursor 3. Due to the symmetry, only half of the molecules were shown. (b) LOL-π analysis of NG1 with isovalue set to 0.52.
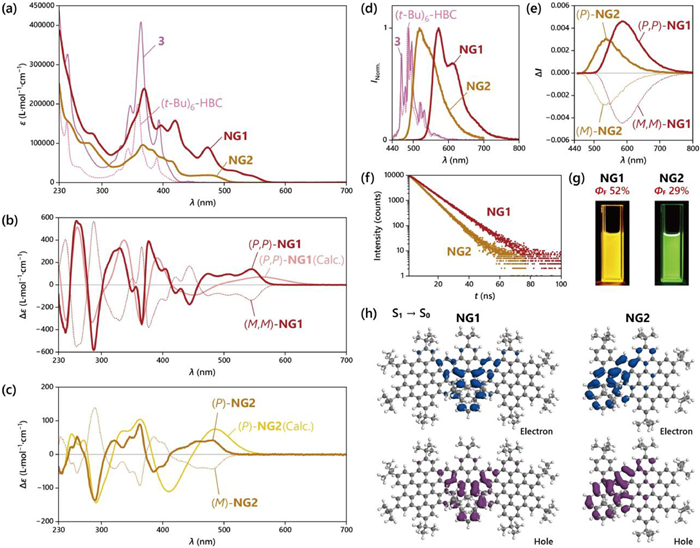
From the UV–vis absorption (Fig. 4a), we found that the uncyclized precursor 3 showed three intense sharp peaks at 393, 364 and 347 nm and two merely observable weak peaks in the visible region at 446 and 438 nm. This absorption pattern resembled closely to (t-Bu)6–HBC (similar sharp characteristic peaks with high intensity at 390, 359 and 343 nm). TD-DFT calculations indicated that these intense peaks were ascribed to the π-π* from the two HBC regions according to the hole-electron distribution analysis localized for the corresponding transitions (Fig. S34 in Supporting information) [69]. When switching to the cyclized product NG1, the absorption curve significantly extended to 585 nm, indicating a much more efficient conjugation between the HBC and the pyrene units. Peaks with moderate absorption coefficients (< 105 L mol−1 cm−1) were found at 546, 518 and 473 nm, these bands were majorly attributed to the first four excitations according to the TD-DFT calculations. According to hole-electron distribution analysis, these bands showed a high involvement of the central peroperylene core with part of HBC units (Fig. S45 in Supporting information) [70,71]. Meanwhile, more intense peaks were found between 460 nm and 310 nm with the absorption maximum at 368 nm (ε = 2.39 × 105 L mol−1 cm−1), these bands were majorly related to the whole NG skeleton.
Figure 4
Figure 4.
(a) UV–vis spectra for NG1 and NG2 with comparison of 4 and (t-Bu)6—HBC. ECD spectra of (b) NG1 and (c) NG2 with comparison of calculated curves. (d) Fluorescence spectra of NG1 (λex = 467 nm), NG2 (λex = 368 nm), 4 (λex = 365 nm) and (t-Bu)6—HBC (λex = 365 nm). (e) CPL spectra, (f) time-resolved fluorescence spectra, (g) photographs under irradiation of 365 nm for NG1 and NG2. (h) Hole-electron analysis for NG1 and NG2 in their optimized first excited states. All the experimental spectra were recorded in CH2Cl2 with concentration c = 5 × 10−6 mol/L.
The enantiomers of optically pure NG1 and NG2 showed mirror-image Cotton effects with intense signals in electronic circular dichroism (ECD) (Figs. 4b and c). The assignment of the absolute configuration was assisted by theoretical calculations. NG1 showed sharp and structured ECD peaks from 585 nm to 230 nm with alternating the sign for 7 times (for (P,P)-NG1, positive at 545, 378, 330 and 260 nm, and negative at 444, 366, 287 and 242 nm). This character was precisely reproduced by TD-DFT calculations. The strongest ECD signal was found at 287 nm (Δε = −575 L mol−1 cm−1) and 260 nm (Δε = +568 L mol−1 cm−1) for (P,P)-NG1. Besides, for the low-energy region, three weaker bands at 545, 507 and 474 nm were found with Δε ~ +100 L mol−1 cm−1, where large dissymmetry factor in electronic absorption (|gabs|) at 9.0 × 10−3 was found at this region. For NG2, the largest |gabs| was found to be 2.4 × 10−3 at 485 nm.
NG1 showed strong orange fluorescence with emission maxima λem at 572 nm and shoulder peaks at 612 and 676 nm (Excitation wavelength λex = 467 nm, Fig. 4d), and the quantum yield ΦF was measured at 52% at this excitation wavelength. Besides, when using the absorption maximum (368 nm) as the excitation wavelength, we obtained a similar ΦF value at 51%. The fluorescence lifetime of NG1 was measured at 10.0 ns according to the time-dependent photoluminescence experiment (Fig. 4f), indicating the radiative transition rate (kr) of 5.1 × 107 s−1, and the nonradiative transition rate (knr) of 4.9 × 107 s−1. In addition, the fluorescence pattern of NG1 exhibited a distinct contrast with the precursor 3 that gave sharp and structured peaks with emission maxima at blue-shifted region (488 nm) and its quantum yield ΦF noticeably decreased to 3%. This comparison indicated that the formation of the two C—C bonds also efficiently enhances the fluorescent properties. According to the calculations, the fluorescence of precursor 3 was majorly attributed to the π-π* transition localized in one of the HBC units (Fig. S49 in Supporting information). This brought precursor 3 a similar fluorescence pattern and quantum yield as (t-Bu)6–HBC (emission maxima at 484 nm with ΦF of 3%). On the contrary, the fluorescence of NG1 was majorly caused by π-π* transition of the central peroperylene core and part of HBC units (Fig. 4h). Similarly, the smaller NG2 showed emission maxima at 519 nm with a quantum yield ΦF of 29% (λex at 368 nm). Furthermore, circularly polarized luminescence was measured for both enantiopure NG1 and NG2. Smaller NG2 gave a CPL dissymmetry factor |glum| value of 3.0 × 10−3 with (P)-enantiomer showing positive sign and (M)-enantiomer negative sign (Fig. 4e). Meanwhile, for NG1, the |glum| value was higher, giving 5 × 10−3 with (P,P)-enantiomer positive sign and (M,M)-enantiomer negative sign. We subsequently calculated the CPL brightness BCPL values [60] of NG1 and NG2. Notably, NG1 showed a considerably high BCPL value up to 305 L mol−1 cm−1 (calculated with λex at 368 nm, Table S2). The BCPL value of NG2 was calculated as 41 L mol−1 cm−1.
In conclusion, we have reported an innovative design strategy for constructing twistacene-embedded chiral NGs containing helicene breaches with high diastereoselectivity. The formed NG1 showed a homochiral feature with flag-hinge-like geometry. Owing to the pyrene core embedded in the NG skeleton, NG1 showed high luminescence with quantum yield ΦF up to 52%. Furthermore, both NG1 and the homologue NG2 showed intense ECD nature and outstanding CPL performance with BCPL values up to 305 L mol−1 cm−1. We thus anticipate this research to provide an innovative concept for designing new types of twisted chiral NGs with fascinating chiroptical properties.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the Zhejiang Provincial Natural Science Foundation, China (No. LY23B040003), and the Science Foundation of Zhejiang Sci-Tech University, China (No. 22062026-Y).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109667.
W. Yang, G. Longhi, S. Abbate, et al., J. Am. Chem. Soc. 139 (2017) 13102–13109. doi: 10.1021/jacs.7b06848
[71]
Y. Hu, G.M. Paternò, X.Y. Wang, et al., J. Am. Chem. Soc. 141 (2019) 12797–12803. doi: 10.1021/jacs.9b05610
Figure 1
(a) Synthetic routes for NG1 and NG2. (b) Single-crystal structure of NG1 with thermal ellopsoids shown at 30% probability (Hydrogen atoms are omitted for clarity). [10]twistacene unit is shown in red. (c) IGMH analysis of NG1 between tert‑butyl groups and central perylene core (Geometry was optimized at the PBE0-D3(BJ)/def2-SVP level of theory). (d) Side-view of single-crystal structure of NG1 with tert‑butyl groups and hydrogen atoms omitted, and packings of (P,P)-NG1 (in red) and (M,M)-NG1 (in green) in the unit cell.
Figure 2
(a) Isomerization process between homochiral and heterochiral NG1 isomers. (b) Cyclization process from 4 to NG1 isomers. The calculations were performed in the PBE0-D3(BJ)/def2-TZVPP//PBE0-D3(BJ)/def2-SVP level, and the Gibbs free energies are given in kcal/mol. Peripheral tert‑butyl groups were omitted for calculation simplicity.
Figure 3
(a) NICS(0) values of NG1 and precursor 3. Due to the symmetry, only half of the molecules were shown. (b) LOL-π analysis of NG1 with isovalue set to 0.52.
Figure 4
(a) UV–vis spectra for NG1 and NG2 with comparison of 4 and (t-Bu)6—HBC. ECD spectra of (b) NG1 and (c) NG2 with comparison of calculated curves. (d) Fluorescence spectra of NG1 (λex = 467 nm), NG2 (λex = 368 nm), 4 (λex = 365 nm) and (t-Bu)6—HBC (λex = 365 nm). (e) CPL spectra, (f) time-resolved fluorescence spectra, (g) photographs under irradiation of 365 nm for NG1 and NG2. (h) Hole-electron analysis for NG1 and NG2 in their optimized first excited states. All the experimental spectra were recorded in CH2Cl2 with concentration c = 5 × 10−6 mol/L.


 DownLoad:
DownLoad:

 下载:
下载:





