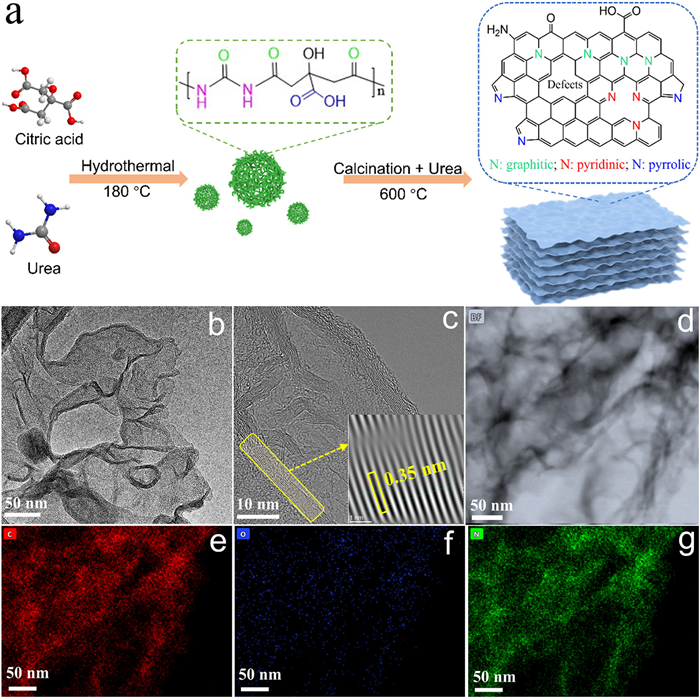
Figure 1.
(a) Illustration of synthesis of metal-free N–C material. (b) TEM, (c) HRTEM, (d) HAADF-STEM, and EDX elemental mapping images including (e) C, (f) O and (g) N of N–C.
Nitrogen-rich carbon for catalytic activation of peroxymonosulfate towards green synthesis
Yu Yao , Jinqiang Zhang , Yantao Wang , Kunsheng Hu , Yangyang Yang , Zhongshuai Zhu , Shuang Zhong , Huayang Zhang , Shaobin Wang , Xiaoguang Duan

Water resources are essential for sustaining life and supporting various human activities, including drinking, agriculture, industry, and ecosystem preservation [1,2]. However, water resources are experiencing severe contamination due to human activities, natural processes, and pollution, leading to significant environmental and health concerns [3]. Therefore, efficient purification of wastewater is crucial to ensure the availability of clean and safe water for present and future generations, as well as to maintain the sustainability of ecosystems and the planet. Among all the technologies for wastewater treatment, peroxymonosulfate (PMS), as an inexpensive oxidant, has shown significant potential for pollutant degradation [4,5]. PMS activation generates highly reactive oxygen species (ROS), such as SO4•− (2.5–3.1 V) and •OH (2.7 V), which contribute to degrading various refractory pollutants in water [6]. However, the practical application of PMS-based oxidation processes faces challenges due to the lack of effective strategies for PMS activation and limited selectivity.
Various energy sources, including thermal energy [7], ultrasound [8], and ultraviolet and visible light [9,10] have been explored to enhance PMS activation. However, the increased energy consumption and higher facility costs limit the large-scale application of PMS oxidation technology. To address this challenge, researchers have used both heterogeneous and homogeneous metal-based catalysts to activate PMS to yield ROS [6]. Nevertheless, metal-containing catalysts would generate metal-containing sludge, leading to potential secondary pollution in aqueous solutions [11,12]. As a result, metal-free catalysts, particularly nitrogen doped carbon (N–C) materials, as sustainable and highly active catalysts, have emerged as green alternatives for PMS activation [13–15]. In addition, the unsaturated coordination environment of nitrogen dopants and structural defects in N–C materials serve as active sites for accelerating PMS-based oxidation processes [12]. However, the synthesis of N–C materials typically requires harsh pyrolysis conditions [14,16], and the resulting low nitrogen doping levels limited their activity in environmental catalysis [17]. Therefore, seeking an economical approach to prepare high-performance N–C catalysts is crucial for developing green and efficient PMS-based advanced oxidation processes.
1,4-Dihydro-2,6-dimethylpyridine-3,5-dicarboxylate (1,4-DHP) finds extensive use in feed additives as well as a hydrogen source in organocatalytic reductive amination and conjugate reduction [18,19]. However, improper handling and discharge of 1,4-DHP may lead to contamination of oceans and rivers, even drinking water, due to its acute toxicity [20,21]. Consequently, significant efforts have been devoted to developing efficient technologies for converting 1,4-DHP into its oxidant of diethyl 2,6-dimethylpyridine-3,5-dicarboxylate (2,6-DHD), an important intermediate used in the synthesis of drugs known as calcium-channel blockers. Photocatalytic technology has garnered increasing attention for selectively oxidizing 1,4-DHP in aqueous solutions due to its sustainability and environmentally friendly attributes. However, photocatalysis is limited by the low sunlight harvesting capability of the semiconductor catalysts and sluggish charge dynamics to initiate redox reactions [22]. Therefore, there is a strong demand for an efficient and clean approach to selectively convert 1,4-DHP into high-value chemicals in a green manner. While the high oxidation ability of PMS in Fenton-like reactions has been recognized, its application in the selective conversion of 1,4-DHP has not yet been reported.
In this work, we synthesized polymer dots (PDs) to induce an epitaxial growth process for N–C materials. The abundant amino groups on the surface of PDs facilitated the growth of urea, leading to the formation of nitrogen-rich N–C materials. Our approach enables the fabrication of N–C materials at a lower temperature of 600 ℃, while achieving a high nitrogen content of 41.8 at%. In contrast, carbon nitride was obtained in direct polymerization of urea without the presence of PDs. The resulting N–C catalyst was applied for PMS activation to selectively oxidize 1,4-DHP. Compared with other carbon-based materials (i.e., carbon nitride, graphene and carbon nanotube), N–C exhibited outstanding oxidation efficiency, while maintaining a high selectivity. Experimental characterization identified the intrinsic ROS are superoxide anion radicals (O2•−) in the N–C/PMS system. This work holds promise for advancing the efficiency and versatility of PMS-based oxidation systems for chemical synthesis.
The synthetic process for the N–C material is illustrated in Fig. 1a. Initially, PDs were synthesized through a hydrothermal method using urea and citric acid as precursors. Transmission electron microscopy (TEM) images show that PDs are uniformly dispersed with an average size of approximately 20 nm (Fig. S1 in Supporting information). Subsequently, the PDs were mixed and copolymerized with urea at 600 ℃. The terminal amino and carboxyl groups on PDs functioned as adsorption sites for urea molecules during pyrolysis, thus facilitating their assembly into nitrogen-doped carbon rings that were fabricated into N–C, instead of carbon nitride.
The morphology of N–C was characterized by TEM imaging, revealing a sheet-like stacking structure with abundant pores (Figs. 1b and c). Additionally, the lattice spacing observed in the high-resolution transmission electron microscopy (HRTEM) image is estimated to be 0.35 nm (insert of Fig. 1c), corresponding to the interlayer stacking distance of graphene [23]. Further analysis through high-angle-annular-dark-field scanning transmission electron microscopy (HAADF-STEM) and element mapping images confirm that N–C is primarily consisted of carbon and nitrogen elements, with trace-level oxygen element (Figs. 1d-g).
X-ray powder diffraction (XRD) patterns were used to study the crystalline structure of all the samples (Fig. 2a). CN exhibited two diffraction peaks at 13.1° and 26.7°, corresponding to the in-plane repeating units of heptazine framework and the interlayer aromatic stacking, respectively [22,24]. However, upon the addition of a certain amount of PDs during the synthesis, the diffraction peak at 13.1° disappeared, and the intensity of the peak at 27.4° diminished and shifted to 26.5°, indicating the formation of a carbon material instead of CN [25,26]. Moreover, N–C displayed a similar peak to N–C(CA+U), but with a lower intensity and a broader width at half-maximum (FWHM) compared to G-plate and M-CNT, suggesting a lower degree of graphitization and a thinner lamellar nanostructure for N–C. These findings align with the TEM images, confirming the porous and 2D thin film structure of N–C. Therefore, it can be concluded that the addition of PDs in the polymerization process leads to the conversion of urea into N-doped carbon rather than CN.
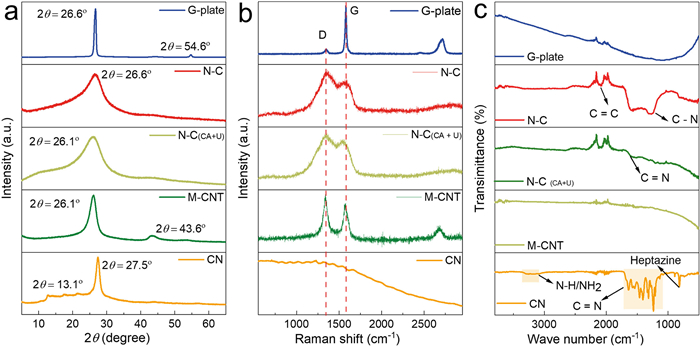
Raman spectra of N–C provided further insights into the microstructural features of N–C, distinguishing it from CN, G-plate, and M-CNT. Two characteristic peaks at 1325 and 1558 cm−1 were observed, ascribing to the crystal defects (D band) and the in-plane stretching sp2 graphitic carbon (G band), respectively (Fig. 2b) [27,28]. Compared with N–C(CA+U), commercial G-plate and M-CNT, N–C exhibited a wider G band and a higher D band, indicating the presence of more carbon defects in the graphitic framework. The intensity ratios (ID/IG) were determined to assess the proportion of non-sp2-hybridized carbons (defective and disordered structures) in the different samples. The values were calculated to be 0.15, 1.07, 1.16 and 1.16 for G-plate, N–C(CA+U), N–C and M-CNT, respectively, meaning that the graphitization degree of N–C was close to that of M-CNT but lower than that of commercial graphene plates [29]. Therefore, the copolymerization of PDs and urea resulted in the generation of N-doped carbon with abundant defects, while CN was obtained in the absence of PDs during urea polymerization.
Fourier transform infrared spectrometer (FTIR) spectra was collected to examine the functional groups present in all the samples (Fig. 2c). CN exhibited characteristic peaks at 809 and 1094 ~ 1635 cm−1, corresponding to the triazine unit and CN heterocycles, respectively, while a broad range of peaks from 3000 cm−1 to 3400 cm−1 indicated the presence of the amino group (N–H). In comparison, N–C(CA+U) displayed peaks at 1094 ~ 1643 cm−1 (C=N) and 2056 ~ 2134 cm−1 (C=C) [30]. Notably, the N–C sample revealed additional functional group peaks at 1256 cm−1 (C–N) [31–33]. These findings indicate that the surface functional groups of the N–C samples differ from those of CN, N–C(CA+U), G-plate, and M-CNT. Thus, the addition of PDs significantly modifies the chemical structures of CN to form a graphitic network that accommodate abundant nitrogen functionalities.
X-ray photoelectron spectroscopy (XPS) analysis was performed to gain further insights into the chemical states of carbon and nitrogen atoms and surface functional groups in the prepared samples. In the CN material, the XPS C 1s spectra (Fig. S2a in Supporting information) exhibited three characteristic peaks at 284.8, 286.2, and 288.2 eV, corresponding to C–C/C=C, C–N, and C–N=C, respectively. In the case of M-CNT and G-plate samples, the peaks at 284.8, 286, and 287 eV was associated with C–C/C=C, C–O, and C=O, respectively. Conversely, the N–C catalyst displayed peaks at 284.8, 286.2, 287, 288.2, 289.66, and 291.3 eV, representing C–C/C=C, C–N, C=O, C–N=C, N–C = O, and π-π transition, respectively [34]. N 1s spectra of CN was deconvoluted into two peaks centered at 398.2 and 401.1 eV, assigning to pyridinic N and graphitic N, respectively. On the other hand, three fitted peaks in the N 1s spectra of N–C were identified, including pyridinic N (398.2 eV), pyrrolic N (399.8 eV), and graphitic N (401.1 eV) (Fig. S2b in Supporting information) [35,36]. This difference in nitrogen species between N–C and CN confirms the formation of N-doped carbon materials when PDs were added during the copolymerization process, instead of triazine and tri-s-triazine rings in carbon nitride. It is worth noting that the N content in N–C reaches as high as 41.8 at%, which is the highest among reported N-doped carbon samples (Figs. S2c and d in Supporting information), confirming the effectiveness of the epitaxial growth strategy employed in this study for obtaining N–C with a high N loading and structural defects.
X-ray absorption fine structure spectroscopy (NEXAFS) measurements were utilized to distinguish the chemical states of C and N between the N–C and CN layers. Fig. 3a shows the C K-edge NEXAFS spectra of N–C and CN, with absorption features observed at 287, 288.3 and 293.8 eV, which can be assigned to the C–N, C–N-C, and C=C/C–C [37,38], respectively. Furthermore, the N K-edge spectrum reveals distinct at 399.3, 401.2, 402.2 and 404.8 eV, corresponding to C–N–C, C=N–C, C–N and N-(C)3, respectively (Fig. 3b) [38,39]. It unequivocally confirms that CN will be transferred to N–C after PD is added to the urea within the same synthesis process. Additionally, solid-state 13C NMR spectroscopy was employed to elucidate the molecular structure of the synthesized carbon materials. In the CN sample, a dominant peak at 164 ppm, assigned to N–C=N in the triazine, was observed (Fig. 3c). Another peak at around 157 ppm is also observed, corresponding to C–N3 in the triazine ring of CN [29]. In contrast, N–C exhibited eight new peaks at 23.4, 48, 64.7, 107, 120, 138, 147.3, and 184.6 ppm, attributed to CH3, C–N, C–O, methylene-pyridine ring, C≡N, C=C, aromatic rings, and C=O [40], respectively. These findings, along with the results from XPS and NEXAFS analyses, strongly support the conclusion that the copolymerization of PDs and urea leads to the formation of nitrogen-doped carbon materials instead of CN. The NMR results are consistent with the observations from XPS analysis, confirming the presence of various oxygen-containing functional groups in the nitrogen-doped carbon materials.
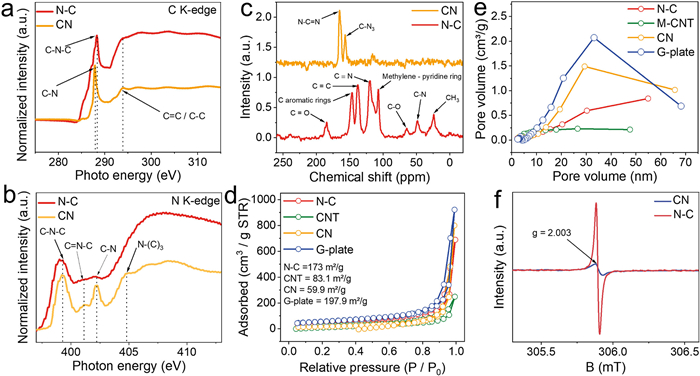
N2 adsorption-desorption isotherms were collected to investigate the porosity and surface area of the prepared samples [28]. Pore size distributions determined by the BJH desorption method Fig. 3d shows typical Ⅳ hysteresis loops, indicating the presence of mesopores in all carbon materials. The corresponding BET specific surface areas (SBET) were measured as 173.0, 197.9, 59.9, and 83.1 m2/g for N–C, G-plate, CN, and CNT, respectively. Fig. 3e revealed that the pore sizes were distributed in the range of 25–35 nm for all samples. Electron paramagnetic resonance (EPR) spectra were acquired to investigate the defective structure of CN and N–C materials. Both samples displayed a great paramagnetic signal at a g factor of 2.003, indicating the presence of unpaired electrons at boundary carbons [24,41]. The peak intensity on N–C was higher compared to CN, attributed to the graphitic network with abundant carbon defects in N–C (Fig. 3f). These findings suggest that the introduction of nitrogen dopants results in the generation of high charge density and creation of structural defects on N–C.
The catalytic performance of the synthesized N–C catalyst was evaluated in the activation of PMS for the selective oxidation of 1,4-DHP. A blank experiment without any catalyst showed that only 8% of 1,4-DHP was oxidized by PMS within 20 min, indicating limited reactivity of the inactivated PMS towards 1,4-DHP oxidation (Fig. 4a and Fig. S3a in Supporting information). When the N–C catalyst was added alone, a removal efficiency of 10% for 1,4-DHP was achieved, demonstrating that the adsorptive ability of N–C catalyst is not significant. However, when PMS and catalysts were co-introduced, remarkable enhancement in 1,4-DHP conversion efficiency was observed. The PMS/N–C system exhibited the highest activity, achieving a 97% conversion within 20 min. In comparison, the efficiency was 29.5% for M-CNT, 17% for CN, 9.2% for G-plate, and 7.7% for N–C(CA+U) in the PMS system (Fig. 4a and Fig. S3b in Supporting information). This also can be observed by. normalized the kinetics rate constant (k) with the specific surface area (SSA) for various catalysts.in Fig. S4 (Supporting information). The selectivity was determined to be nearly 100% for N–C, and above 90% for M-CNT, CN, G-plate, and N–C(CA+U) samples (Fig. 4a, and Figs. S5 and S6 in Supporting information).
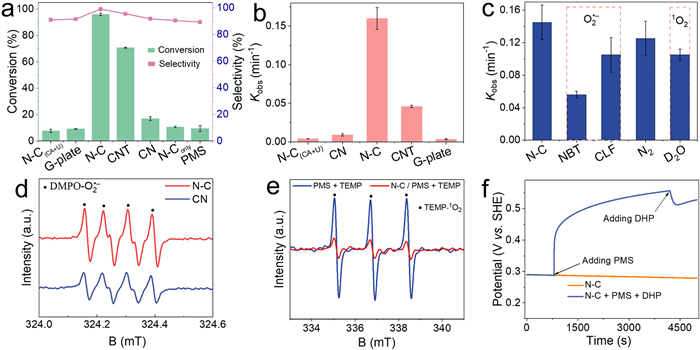
Additionally, N–C exhibited the highest first-order kinetic rate constant of 0.16 min−1, while CNT, CN, G-plate, and N–C(CA+U) only attained rate constants of 0.046, 0.009, 0.0035, and 0.0042 min−1, respectively (Fig. 4b). These results indicate that both pure carbon and carbon nitride exhibit lower catalytic activity in PMS activation for 1,4-DHP oxidation compared to the N–C catalyst. It is further demonstrated that the N–C catalyst exhibits high efficiency in the PMS system for the oxidation of 1,4-DHP, primarily due to the increased number of N dopants and structural defects [15,42,43]. These results strongly confirm that the N-dopant and defects present in carbon skeleton play crucial roles in activating PMS to coordinate selective oxidation, thus enhancing conversion efficiency and keeping high selectivity of 1,4-DHP.
Quenching experiments were conducted to investigate the ROS in the activation of PMS process for the selective oxidation of 1,4-DHP using the N–C catalyst. It is known that hydroxyl •OH, persulfate (SO5•−) and SO4•− radicals are typically generated during the PMS activation process [44,45]. However, in this study, •OH, SO5•− and SO4•− radicals were quenched with the presence of large amounts of ethanol in the solvent [46,47]. Therefore, the contribution of •OH, SO5•− and SO4•− to the transformation of 1,4-DHP can be ignored in this system. Other potential ROS such as single oxygen (1O2) or O2•− were considered for the oxidation of 1,4-DHP, and specific ROS quenching agents were introduced into the reaction system to distinguish their contributions. Nitro blue tetrazolium (NBT) and chloroform (CLF) were separately added to capture O2•− in the N–C/PMS system (Fig. S7a in Supporting information) [47,48]. Fig. 4c demonstrated that the conversion efficiency is significantly inhibited upon the addition of NBT and CLF into the N–C/PMS system. With the addition of NBT and CLF, the first-order kinetic rate constant decreased to 0.056 and 0.105 compared with the control group of 0.16 min−1, respectively. Thus, O2•− was generated and participated in 1,4-DHP oxidation. Furthermore, deuteroxide (D2O) solvent was used as a replacement for water to investigate the contribution of 1O2 in the reactive system. This substitution of water by D2O can significantly extend the lifetime of 1O2 in N–C/PMS aqueous system, and thus boost singlet oxygenation reactions [42,49]. Results show that the use of D2O as a solvent in the N–C/PMS system impeded the conversion of 1,4-DHP (Fig. S7b in Supporting information), suggesting that the contribution of 1O2 was insignificant to the conversion of 1,4-DHP in the N–C/PMS system. The ROS capturing experiment by EPR in Fig. 4d exhibit characteristic peaks corresponding to 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-O2•− with an intensity ratio of 1:1:1:1 [10,48], indicating the production of O2•− in the system. Furthermore, 2,2,6,6-tetramethyl-4-piperidinol (TEMP) was selected as a spin trapping agent to provide further verification of the presence of TEMP-1O2 in the N–C/PMS system. Interestingly, in the PMS only system (no catalyst), a stronger triplet signal with an intensity ratio of 1:1:1 can be observed in Fig. 4e [50,51]. However, the triplet signal is remarkably reduced with the presence of NC. The results indicate that the generation of 1O2 in the N–C/PMS system primarily originates from PMS rather than by N–C-induced catalysis. Therefore, based on these results, it can be concluded that O2•−, rather than 1O2, is the primary ROS accounting for 1,4-DHP conversion within the system.
To further explore the electron transfer process in the PMS activation process, open-circuit potentials were measured using the N–C catalysts. The initial potential of N–C was approximately 0.29 V without the addition of PMS, and it rose to around 0.56 V after the introduction of PMS (Fig. 4f). Thus, PMS can interact with the N–C catalyst to produce metastable intermediates (N–C/PMS), subsequently raising the oxidation potential of carbon surface and initiating an electron transfer process (ETP). The potential decreased from 0.56 V to 0.5 V after the addition of 1,4-DHP, validating that the complex intermediates abstract electrons from the adsorbed 1,4-DHP to realize the oxidation [43,52,53].
To investigate the influence of ROS in the PMS system, a controlled experiment was conducted to eliminate DO (dissolved oxygen) by bubbling nitrogen [43,54]. The kinetic constant was reduced to 0.12 min−1 (Fig. 4c), implying that DO is responsible for the generation of O2•− in the N–C/PMS. However, the amount of generated 2,6-DHD remained nearly constant under both aerobic and anaerobic conditions, indicating that O2•− did not participated in the formation of 2,6-DHD (Fig S8 in Supporting information). In the ETP pathway, the polarized nitrogen sites and carbon defects in N–C would complex with HSO5•− to form a metastable intermediate which will elevate the carbon potential with an electron withdrawing capacity toward vulnerable organics [8,55]. Then, 1,4-DHP acts as an electron donor and undergoes transformation into a 1,4-DHP radical via electron transfer. The 1,4-DHP radical would donate electrons to DO and HSO5•−, resulting in the formation of O2•−, sulfate ions, and 1,4-DHP cation. Also, O2•− may have a small contribution to initial 1,4-DHP oxidation and convert to hydrogen peroxide. Ultimately, the generated 1,4-DHP cation will experience internal electrons transfer in accompany with deprotonation to produce 2,6-DHD (Fig. 5).
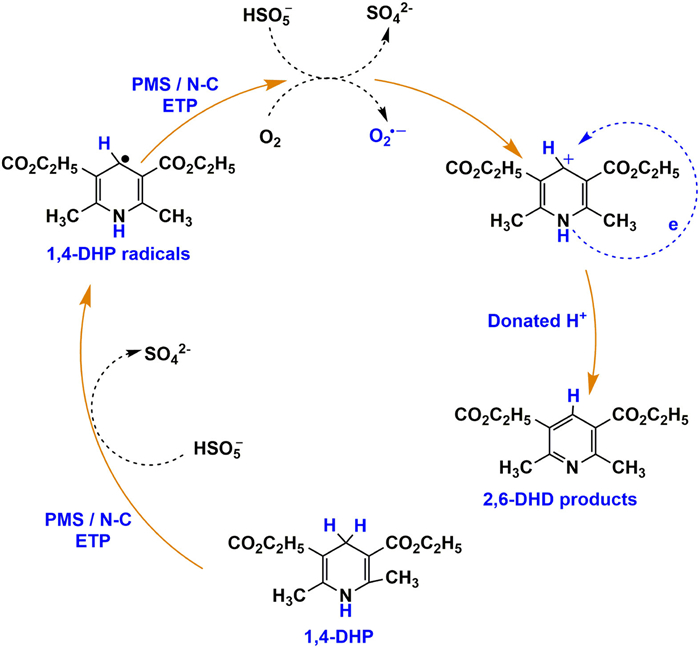
In summary, we utilized a facile polymer dot-assisted approach to synthesize nitrogen-rich carbon materials at a relatively low temperature. Characterization results confirmed that urea grew along the functional groups on the surface of PDs to form N-rich carbons. The resulting N–C material exhibited abundant N dopants and structural defects, which demonstrated superior conversion efficiency and selectivity for 1,4-DHP oxidation in the PMS activation system. Quenching experiments and EPR measurements collectively elucidated the crucial role of electron transfer process in the N–C/PMS system induced by nitrogen dopants and defects as well as the \ function of O2•− in 1,4-DHP transformation. This work provides comprehensive insights into the fabrication of highly active N–C materials for catalytic oxidation and green synthesis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors acknowledge the financial support from Australian Research Council under DECRA Scheme (No. DE210100253) and merit SXR beamline from Australian Nuclear Science and Technology Organization (ANSTO, No. AS231/SXR/19680).
Supplementary material associated with this article can be found, in the online version, at doi:
Y.F. Huo, M. Yang, M.Y. Lei, et al., Water Environ. J. 38 (2024) 162–168. doi: 10.1111/wej.12895
B.R. Scanlon, S. Fakhreddine, A. Rateb, et al., Nat. Rev. Earth Env. 4 (2023) 87–101. doi: 10.1038/s43017-022-00378-6
A. Mukhopadhyay, S. Duttagupta, A. Mukherjee, J. Environ. Chem. Eng. 10 (2022) 107560. doi: 10.1016/j.jece.2022.107560
X.G. Duan, Z.M. Ao, D.G. Li, et al., Carbon 103 (2016) 404–411. doi: 10.1016/j.carbon.2016.03.034
X.G. Duan, H.Q. Sun, M. Tade, et al., Catal. Today 307 (2018) 140–146. doi: 10.1016/j.cattod.2017.04.038
X.G. Duan, C. Su, J. Miao, et al., Appl. Catal. B: Environ. 220 (2018) 626–634. doi: 10.1016/j.apcatb.2017.08.088
J.L. Wang, S.Z. Wang, Chem. Eng. J. 334 (2018) 1502–1517. doi: 10.1016/j.cej.2017.11.059
W.Y. Han, D.G. Li, M.Q. Zhang, et al., J. Hazard. Mater. 395 (2020) 122695. doi: 10.1016/j.jhazmat.2020.122695
A.B. Alayande, S. Hong, Environ. Pollut. 307 (2022) 119513. doi: 10.1016/j.envpol.2022.119513
G. Nie, L. Xiao, J.X. Bi, et al., Appl. Catal. B: Environ. 315 (2022) 121584. doi: 10.1016/j.apcatb.2022.121584
X.G. Duan, C. Su, L. Zhou, et al., Appl. Catal. B: Environ. 194 (2016) 7–15. doi: 10.1016/j.apcatb.2016.04.043
X.G. Duan, S. Indrawirawan, H.Q. Sun, et al., Catal. Today 249 (2015) 184–191. doi: 10.1016/j.cattod.2014.10.005
X.G. Duan, H.Q. Sun, Z.M. Ao, et al., Carbon 107 (2016) 371–378. doi: 10.1016/j.carbon.2016.06.016
S.H. Ho, Y.D. Chen, R.X. Li, et al., Water Res. 159 (2019) 77–86. doi: 10.1016/j.watres.2019.05.008
X.G. Duan, Z.M. Ao, H.Q. Sun, et al., ACS Appl. Mater. Interfaces 7 (2015) 4169–4178. doi: 10.1021/am508416n
W.J. Tian, H.Y. Zhang, X.G. Duan, et al., Adv. Funct. Mater. 30 (2020) 1909265. doi: 10.1002/adfm.201909265
H.R. Li, J.Y. Tian, Z.G. Zhu, et al., Chem. Eng. J. 354 (2018) 507–516. doi: 10.1016/j.cej.2018.08.043
X.J. Wei, L. Wang, W.L. Jia, et al., Chin. J. Chem. 32 (2014) 1245–1250. doi: 10.1002/cjoc.201400521
K.K. Borowicz, M. Gasior, Z. Kleinrok, et al., Eur. J. Pharmacol. 323 (1997) 45–51. doi: 10.1016/S0014-2999(97)00020-4
S.D. Dhengale, V.M. Naik, G.B. Kolekar, et al., Res. Chem. Intermed. 47 (2021) 3263–3287. doi: 10.1007/s11164-021-04473-z
H. Saffarian, F. Karimi, M. Yarie, et al., J. Mol. Struct. 1224 (2021) 129294. doi: 10.1016/j.molstruc.2020.129294
J.Q. Zhang, X.H. An, N. Lin, et al., Carbon 100 (2016) 450–455. doi: 10.1016/j.carbon.2016.01.027
S. Indrawirawan, H.Q. Sun, X.G. Duan, et al., J. Mater. Chem. A 3 (2015) 3432–3440. doi: 10.1039/C4TA05940A
J.Q. Zhang, Y.G. Li, X.L. Zhao, et al., Nano Energy 89 (2021) 106357. doi: 10.1016/j.nanoen.2021.106357
Y.W. Gao, T. Li, Y. Zhu, et al., J. Hazard. Mater. 393 (2020) 121280. doi: 10.1016/j.jhazmat.2019.121280
Y.W. Gao, Z.H. Chen, Y. Zhu, et al., Environ. Sci. Technol. 54 (2020) 1232–1241. doi: 10.1021/acs.est.9b05856
W. Zhang, Y. Li, X.B. Fan, et al., Carbon 155 (2019) 268–278. doi: 10.1016/j.carbon.2019.08.071
P. Liang, C. Zhang, X.G. Duan, et al., Environ. Sci. Nano 4 (2017) 315–324. doi: 10.1039/C6EN00633G
J. Li, Y. Zhang, X. Zhang, et al., ACS Appl. Mater. Interfaces 7 (2015) 19626–19634. doi: 10.1021/acsami.5b03845
F. Li, M.E. Han, Y. Jin, et al., Appl. Catal. B: Environ. 256 (2019) 117705. doi: 10.1016/j.apcatb.2019.05.007
Q.F. Li, C. Ren, C.T. Qiu, et al., Chin. Chem. Lett. 32 (2021) 3463–3468. doi: 10.1016/j.cclet.2021.05.039
G.Q. Zhang, Y.S. Xu, G.S. Liu, et al., Chin. Chem. Lett. 34 (2023) 107383. doi: 10.1016/j.cclet.2022.03.106
Y. Zhang, L. Wu, S. Wang, et al., Chin. Chem. Lett. 35 (2023) 108551. doi: 10.1016/j.cclet.2023.108551
W.J. Tian, J.K. Lin, H.Y. Zhang, et al., J. Hazard. Mater. 408 (2021) 124459. doi: 10.1016/j.jhazmat.2020.124459
D.G. Li, X.G. Duan, H.Q. Sun, et al., Carbon 115 (2017) 124459.
X. Chen, X.G. Duan, W.D. Oh, et al., Appl. Catal. B: Environ. 253 (2019) 419–432. doi: 10.1016/j.apcatb.2019.04.018
H. Jiang, J.X. Gu, X.S. Zheng, et al., Energ. Environ. Sci. 12 (2019) 322–333. doi: 10.1039/C8EE03276A
Y. Zheng, Y. Jiao, Y. Zhu, et al., Nat. Commun. 5 (2014) 3783. doi: 10.1038/ncomms4783
Y. Xu, M. Fan, W. Yang, et al., Adv. Mater. 33 (2021) e2101455. doi: 10.1002/adma.202101455
T. Wang, Y. Sun, L. Zhang, et al., Adv. Mater. 31 (2019) e1807876. doi: 10.1002/adma.201807876
S.J. Wang, L. Chen, X.L. Zhao, et al., Appl. Catal. B: Environ. 278 (2020) 119312. doi: 10.1016/j.apcatb.2020.119312
W. Ren, G. Nie, P. Zhou, et al., Environ. Sci. Technol. 54 (2020) 6438–6447. doi: 10.1021/acs.est.0c01161
J. Wang, X.G. Duan, J. Gao, et al., Water Res. 185 (2020) 116244. doi: 10.1016/j.watres.2020.116244
P.P. Zhang, Y.Y. Yang, X.G. Duan, et al., ACS Catal. 11 (2021) 11129–11159. doi: 10.1021/acscatal.1c03099
W.T. Zheng, Y.B.A. Liu, W. Liu, et al., Water Res. 194 (2021) 116961. doi: 10.1016/j.watres.2021.116961
Y.Y. Yang, P.P. Zhang, K.S. Hu, et al., Appl. Catal. B: Environ. 286 (2021) 119903. doi: 10.1016/j.apcatb.2021.119903
H. Wu, X.Y. Xu, L. Shi, et al., Water Res. 167 (2019) 115110. doi: 10.1016/j.watres.2019.115110
Y.D. Chen, X.G. Duan, X. Zhou, et al., Chem. Eng. J. 409 (2021) 128207. doi: 10.1016/j.cej.2020.128207
P.H. Shao, Y.P. Jing, X.G. Duan, et al., Environ. Sci. Technol. 55 (2021) 16078–16087. doi: 10.1021/acs.est.1c02042
Y.B. Xie, Y. Liu, Y.J. Yao, et al., Chin. Chem. Lett. 33 (2022) 1298–1302. doi: 10.1016/j.cclet.2021.07.055
Y. Liu, F.X. Cun, D.Q. Tian, et al., J. Hazard. Mater. 435 (2022) 128975. doi: 10.1016/j.jhazmat.2022.128975
W. Ren, C. Cheng, P.H. Shao, et al., Environ. Sci. Technol. 56 (2022) 78–97. doi: 10.1021/acs.est.1c05374
W. Ren, L.L. Xiong, X.H. Yuan, et al., Environ. Sci. Technol. 53 (2019) 14595–14603. doi: 10.1021/acs.est.9b05475
Y.X. Wang, X. Li, S.N. Liu, et al., ACS Catal. 12 (2022) 2770–2780. doi: 10.1021/acscatal.1c05447
W. Han, D. Li, Y. Kong, et al., J. Colloid Interface Sci. 646 (2023) 633–648. doi: 10.1016/j.jcis.2023.05.092

Figure 1 (a) Illustration of synthesis of metal-free N–C material. (b) TEM, (c) HRTEM, (d) HAADF-STEM, and EDX elemental mapping images including (e) C, (f) O and (g) N of N–C.

Figure 2 Catalyst characterizations. (a) XRD patterns, (b) Raman spectra, and (c) FTIR spectra for CN, N–C, M-CNT, N–C(CA+U) and G-plate.

Figure 3 (a) C and (b) N K-edge NEXAFS of N–C and CN. (c) Solid 13C NMR results for CN and N–C. (d) N2 adsorption and desorption isotherms and (e) pore size distributions for N–C, CNT, CN and G-plate. (f) Solid EPR spectra for CN and N–C.

Figure 4 Performance evaluation. (a) Conversion and selectivity for CN, N–C, M-CNT, N–C(CA+U) and G-plate in selective oxidation of 1,4-DHP. (b) Kinetic rate constant of selective oxidation of 1,4-DHP (25 ppm) with various carbon catalyst in the PMS system. (c) Trapping experiments with different scavengers in PMS activation for oxidation of 1,4-DHP using N–C as the catalyst. Reaction conditions: [Catalysts]0 = 5mg/L, [1,4-DHP]0 = 25 mg/L, [PMS]0 = 0.2 mmol/L, [CLF]0 = 20 mmol/L, [NBT]0 = 1 mmol/L. EPR measurements for monitoring (d) O2•− (DMPO) and (e) 1O2 (TEMP) in PMS activation process for the oxidation of 1,4-DHP. (f) Open-circuit potential curves on N–C electrode in PMS mediated oxidation system.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
