
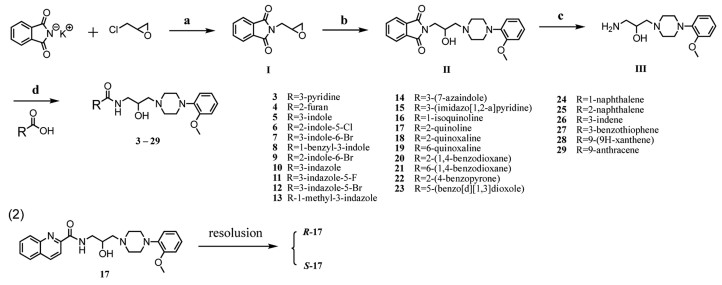
Scheme 1.
Modification strategy of target compounds in present study.
Discovery of an enantiopure N-[2-hydroxy-3-phenyl piperazine propyl]-aromatic carboxamide derivative as highly selective α1D/1A-adrenoceptor antagonist and homology modelling
Junjun Huang , Ran Chen , Yajian Huang , Hang Zhang , Anran Zheng , Qing Xiao , Dan Wu , Ruxia Duan , Zhi Zhou , Fei He , Wei Yi

Benign prostatic hyperplasia (BPH) is an abnormal increase in the size of the prostate gland due to unregulated hyperplasia growth of both epithelial and stromal cells of the prostate in the transitional zone surrounding the urethra [1,2]. BPH is a frequent cause of lower urinary tract symptoms (LUTS) in men and is a common benign neoplasm particularly in ageing men, the prevalence of BPH apparent in around 50% of men who are above 50 years, and 90% who are above 80 years old [3]. BPH/LUTS causes problems with micturition, including increased frequency, due to outflow obstruction that has a dynamic component due to α1-adrenoceptors mediated contraction of the bladder back, prostate and urethra [4].
α1-Adrenergic receptors (ARs) belong to G protein coupled receptor (GPCR) superfamily and coupled with Gq/11 protein involving phospholipase C activation, followed by the increase of intracellular calcium levels. In the late 1980s, molecular biology studies identified three α1-AR subtypes, classified as α1A, α1B and α1D [5]. α1A plays a dominant role in controlling human prostatic smooth muscle contraction. Many α1A-AR subtype selective antagonists have since been discovered, including silodosin [6], however, α1A-selective compounds have not been proven to be very effective in relieving LUTS, especially the symptom of irritation. Subsequent research discovered that α1D was involved in the mediation of LUTS, and the blocking of α1D relaxes the detrusor muscles of the bladder which prevents storage symptoms, thus, additional α1D antagonism may be useful, particularly in prostatic hyperplasia [7,8]. On the other hand, antagonism of the α1B-AR will lead to cardiovascular side effects, such as hypotension, as α1B expressed predominantly in the heart and vascular smooth muscle [9,10]. Collectively, antagonists with better selectivity for α1A and α1D over α1B-AR may alleviate the symptoms associated with BPH efficiently without causing significant cardiovascular side effects. Unfortunately, few ligands concomitantly recognize both α1A and α1D subtypes and rule out α1B, such a lack of selectivity limits their administration in vivo and their therapeutic use [11].
Naftopidil (NAF, 1) is a phenylpiperazine-based α1D/1A-AR antagonist used for treating BPH/LUTS in clinic [8]. NAF has approximately 3- and 17-fold higher potency for the α1D-AR than for the α1A- and α1B-AR subtypes, respectively [12]. Our lab has engaged in the research of NAF and similar α1 subtype blockers for years [13-17]. We previously synthesized phenylpiperazine related structures with amide and substituted indole ring (representative compound 2, Scheme 1), which exhibited moderate α1D/1A-AR selectivity and satisfied anti-BPH activities in vivo [16,17], however, systematic structure–activity relationship (SAR) of this o-methoxyphenyl piperazine with carboxamide skeleton is still limited. Kuo et al. reported several structures of phthalimide-phenylpiperazines with 2-hydroxypropanol linker [18], whereas they only studied the substitution effect on phenyl ring which connected with phthalimide and without in vivo study yet.
Based on the recent resolved α1B crystal structure bound with inverse agonist (+)-cyclazosin [19], a large proportion of aromatic residues were involved in binding pocket, and most of these amino acids were conserved in both α1A and α1D -ARs. This might also explained the proposed ligand pharmacophore model of α1-ARs, in which one or two hydrophobic aromatic groups (HAR) were indispensable for maintaining activity [20,21]. Therefore, we herein replaced indole moiety of compound 2 with fused aromatics or various hetero cyclic rings (Scheme 1) in order to systematically study the effect of aromatics on the pharmacological profile. On the other hand, as we know that the degree of structure diversity in the left hand side was greater than that in the right hand side of molecule, we reserved o-methoxy phenylpiperazine group [11,22]. In addition, we also changed propane linkage into 2-hydroxypropanol to improve the flexibility of overall molecules. By utilizing above strategies, twenty-seven N-[2-hydroxy-3-phenylpiperazinepropyl]-aromatics carboxamide derivatives were designed and synthesized; highly selective and potent α1D/1A-subtype derivatives were expected. As a result, new surrogate 17 with quinoline ring was found to barely exhibit any affinity with α1B-AR, and certainly performed a remarkable subtype selectivity towards α1A- and α1D-ARs. Chiral pharmacological behaviors of two enantiomers R-17 and S-17 were also investigated in rat BPH model, and R-configuration was found to behave as preferentially active enantiomer compared with its antipode. Solid SAR and computer-aided modelling study was also discussed in this research.
According to Scheme 1, mono-, double- and triple aromatic rings were considered as isosteres of naphthalene and were introduced to examine the influence of group size. Besides, aromatic rings with heteroatoms, such as O, N and S have quite different character and interaction potential with the binding site. Therefore, target compounds 3–29 were designed and shown in Scheme 2 [18]. Epoxide Ⅰ was formed by substitution reaction with epichlorohydrin, followed by ring opening reaction with o-methoxy phenyl piperazine in 2-propanol to yield intermediate Ⅱ. Intermediate Ⅱ was then converted into prime amine Ⅲ by deprotecting the phthalimide groups with hydrazine hydrate. The amine Ⅲ was then coupled with various carboxylic acids using hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) to give the desired products 3–29.
All target compounds were evaluated for their biological activities toward α1A-AR using calcium mobilization assays [9,10]. The initial screening was carried out at a concentration of 10 µmol/L for each compound, and most of compounds displayed 90%–100% inhibition, so they were all further evaluated for their half-maximal inhibitory concentration (IC50) values toward α1A-AR (Table 1). Among all compounds, mono-ring substitution 3 and 4 showed the worst inhibitory activity (IC50 > 300 nmol/L). Indole substitution analogues 5–9 exhibited moderate antagonistic activity (IC50 > 100 nmol/L), while indazole derivatives 10–13 displayed ten times higher potency than indole ring (IC50 = 27.50–73.77 nmol/L). 7-Azaindole 14 and imidazo[1,2-a]pyridine 15 also performed comparable potency to indazole. Analogues 16 and 17 owned very small structural differences, however, introduction of quinoline (compound 17, IC50 = 24.82 nmol/L) improved ten-fold higher activity compared with isoquinoline analogue (IC50 = 267.4 nmol/L), indicating the significance of the substitution position of overall aromatic ring. The following quinoxaline derivative 18 remained the same activity (IC50 = 30.23 nmol/L), which also supported that 2-substitution was preferred for α1A receptor recognition, as 6-substituted quinoxaline derivative 19 led to a sharp reduction in potency (IC50 = 146.3 nmol/L). The same trend could also be observed on 1,4-benzodioxane compounds 20 and 21 (IC50 = 31.07 nmol/L vs. 82.02 nmol/L). In case of triple ring analogues, 9-xanthene derivative 28 showed almost equal activity as compared with tamsulosin (IC50 = 3.537 nmol/L), however, 9-anthracene substitution 29 resulted in a 35-times decrease of activity (IC50 = 122.5 nmol/L).
Overall, the introduction of hetero atoms into aromatic rings tend to improve the antagonistic affinity toward α1A-AR, this might due to the hetero atoms behaved as hydrogen bond acceptor (HBA) group [21]. Meanwhile, multi-ring substitution exhibited satisfied antagonistic affinity compared with mono-aromatic derivatives, according to this, the pocket interacting this fragment might be characterized by the presence of more aromatic amino acid residues. A significant dependency on substituent position was also observed, which supported that distances and angles between pharmacophore features matter the magnitude of activity [23].
Next, top two promising α1A- antagonist compounds (17 and 28, IC50 < 25 nmol/L) were selected to be further tested for their inhibitory activities toward α1B- and α1D-ARs. As shown in Table 2, quinoline analogue 17 behaved as α1A/1D- selective inhibitors, whereas compound 28 tend to display higher affinity toward α1A and α1B- subtypes than α1D, probably due to the preference for recognition of bulky xanthene group by α1B- receptor. Compound 17 aroused our great attention that exhibited very weak antagonistic affinity on α1B-AR (IC50 = 7501 nmol/L), and salient subtype selectivity mainly toward α1A- and to a minor extent on α1D-AR (α1A/α1B = 302.22, α1D/α1B = 16.32), exceeding reference drugs. Therefore, rat tissue based functional assay will be carried out to ensure their activity and selectivity.
The antagonistic effects of compound 17 and 28 on Sprague-Dawley rat prostatic vas deferens (α1A), spleen (α1B), and thoracic aorta (α1D) were characterized to assess the sub-receptor selectivity of the compounds (Table 3). NAF mainly displays selectivity for α1D- and, to a minor extent, for α1A- with respect to α1B -AR. Tamsulosin is a high affinity antagonist at functional α1-ARs with a selectivity α1D ≥ α1A > α1B [24]. Silodosin is significantly more selective for α1A abundant prostate tissue compared with other α1-AR subtypes [6]. The inhibitory activity toward α1A-AR of compound 17 and 28 was comparable to that of NAF, while, they improved the antagonistic activities on α1D -AR (pA2 > 8 vs. 7.93 of NAF). Both of these testing molecules displayed similar pA2 values on α1A- (7.5 < pA2 < 8) or α1D-ARs (8 < pA2 < 9) and much higher subtype selectivity toward α1A- and α1D- ARs than reference drugs. We noted that the antagonist affinities toward α1A- and α1D in tissue-based functional assay diverged from cell-based calcium assay (Table 2), representing different trends, as testing compounds mainly blocked α1A in calcium assay while they exhibited stronger inhibition on α1D in tissue-based assay. This phenomenon might due to two different assessment systems between a recombinant receptor in cells and a native receptor in animal tissues [25,26]. With respect to α1B, it is worth noting that compound 17 almost lost affinity on α1B -AR (pA2 < 5.5), which is consistent with the result of calcium assay, indicating that 17 might be a promising candidate for the treatment of BPH with high subtype selectivity.
Given the robust potential of compound 17 as a highly selective α1A/1D-antagonist, we further evaluated the anti-BPH effects of 17 in estrogen/androgen (E/T)-induced rat BPH model in vivo. The animal use and care protocols were reviewed and approved by the Ethics Committee of Guangzhou Medical University (approval number GY2022-208). Considering one chiral center was involved in the structure of 17, two enantiomers might exert different pharmacological activities, therefore, R-17 and S-17 were separated by preparative HPLC using chiral column. The absolute configurations were identified by Mosher ester analysis (Figs. S1–S3 in Supporting information).
In the BPH model group, the values of wet weight, wet weight index, volume and volume index of the rat prostate were significantly larger than those in the sham operated group (Table 4, P < 0.001). Positive drug NAF (10.0 mg/kg) exhibited moderate effects on inhibiting both wet weight and wet weight index but little impact on prostate volume reduction, which was consistent with previous results [13,16]. Low dose (LOW, 2.0 mg/kg) of both R-17 and S-17 could not inhibit the progress of BPH, however, the situation was quite different when high dose (HIG, 5.0 mg/kg) administrated: R-17 clearly decreased the wet weight index and volume index of hyperplastic prostate (P < 0.001), while S-17 performed negligible effect on hyperplastic prostate.
The optical microscopy observations of hematoxylin–eosin-stained samples were compared, as shown in Fig. 1a. The sham operated group maintained the size and shape of the acinar gland well without atrophy. In the BPH model group, the glandular epithelium of prostate acini showed uneven hyperplasia; the contents of the lumen were increased, and the epithelial cells were markedly thickened. Administration of R-17 and S-17 displayed discrepancy: the prostatic acini apparently returned to normal with clear arrangement and uniform size in R-17 HIG group, while S-17 does not show obviously inhibitory activity. Quantitative analysis also supported that 5.0 mg/kg of R-17 greatly suppressed the development of BPH with the minimum thickness of epithelium in rat prostate (P < 0.001 compared with model control group, P < 0.001 compared with S-17 HIG group).
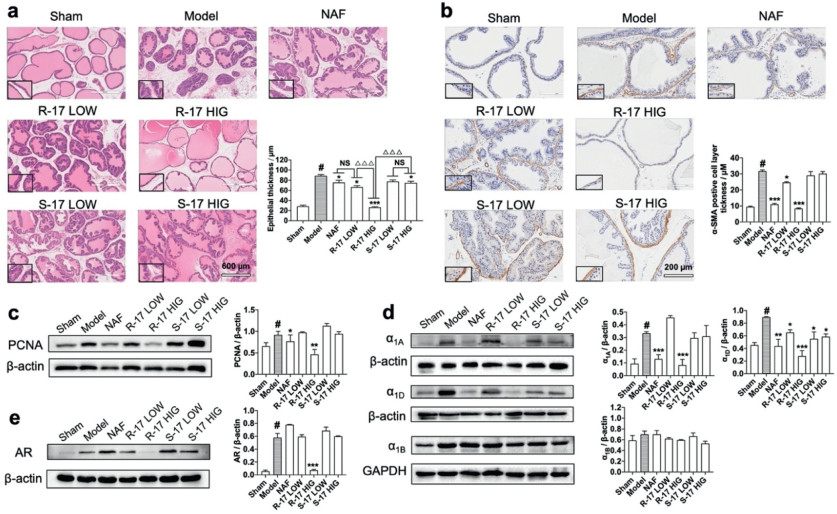
Smooth muscle alpha-actin (α-SMA) is a microfilament protein with contractile properties that is widely distributed in stromal smooth muscle in BPH human prostate [27]. In the BPH model, the acinus was surrounded with α-SMA positive smooth muscle layer compared with that in the sham operated group (Fig. 1b). Administration of R-17 groups reduced thickness of the SMA-positive layer, and the most significant reduction was obtained at a high dose of R-17, indicating R-17 could inhibit the extent of hyperplasia in stromal tissue in BPH to an extent. In addition, PCNA was used as a proliferation marker in rat prostate [28], and R-17 HIG treatment obviously exhibited a decrease in PCNA expression (Fig. 1c, P < 0.01), while S-17 did not show any effect. To estimate the effect of R-17 and S-17 on α1-adrenoceptor subtypes in vivo, we analyzed the expression of α1A, α1D, and α1B protein in prostate tissue by Western blot analysis. As shown in Fig. 1d, the BPH group showed an increase in α1A and α1D expression compared with the sham-operated group (P < 0.05). The α1-AR expression levels in the NAF-treated group were 0.13 ± 0.01 (α1A, P < 0.001) and 0.43 ± 0.02 (α1D, P < 0.01), which were lower than those in the BPH group. Low- and high-dose of S-17 groups showed no effect on α1A (P > 0.05), while exhibited decreased expression on α1D (P < 0.05). Apparently, the high-dose R-17 group produced remarkable reduction on both α1A and α1D (P < 0.001). Regarding of α1B, no significant change was observed in all groups. We also executed Western blot on androgen receptor (AR) and only R-17 HIG group strongly downregulated the AR expression in rat prostate tissue (Fig. 1e).
We are interested in the superior receptor selectivity of R-17, specifically, the unique behavior of low antagonistic affinity toward α1B-AR. Although several subtype-selective ligand pharmacophore models and quantitative structure–activity relationship (QSAR) studies have been reported [21,23,29], this prediction greatly depend on structural diversity of training set compounds and model selection process. Herein, we would like to rationalize the receptor selectivity between α1A and α1B-AR by computer-aided modelling study.
Initially, the α1A homology model (Fig. S4 in Supporting information) was constructed based on the sequence of α1B (PDB id: 7B6W, resolution: 3.1 Å, co-crystallized with its inverse agonist (+)-cyclazosin) [19], the crystal structure of which was reported in 2022 and shares a relatively high BLAST identity of 63% with human α1A. The rational binding pattern of compound R-17 in complex with α1A was carried out through multiple steps including homology modelling, induced-fit docking, molecular dynamics and clustering by Maestro (Schrödinger, LLC, New York, NY). The resulting five representative models which were generated after clustering based on ligand RMSD and SIFt (Structural interaction fingerprints) exhibited quite different ligand-binding conformations (Fig. S5 in Supporting information), and also different protein-ligand interaction profiles, therefore, 100 ns molecular dynamics (MD) was conducted for each model in order to examine the stability (Table S1 in Supporting information). Finally, Model 3 was selected to be a convincing binding pose shown in Fig. 2a and Fig. S6a (Supporting information).
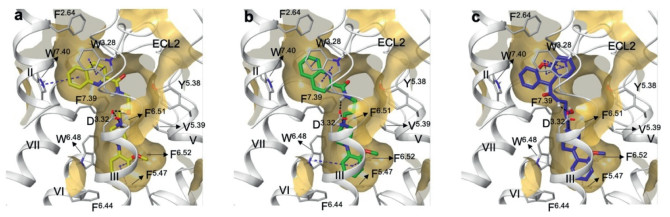
Compound R-17 was fitted nicely into the 7-shaped binding pocket of α1A, which contains two aromatic (AR) centers on each side of ligand and one positive ionizable (PI) nitrogen of piperazine moiety (Fig. S6a). Two aromatic ring segments at both ends of R-17 were shown: quinoline ring was positioned in a upper aromatic pocket, making aromatic-aromatic edge-to-face with W3137.40 (the superscript refers to the Ballesteros-Weinstein numbering) and aromatic-aromatic stacking interaction with W1023.28; on the other side, o-methoxyphenyl ring was positioned in a lower aromatic pocket, making pi-pi stacking with F2896.52 and F1935.47, and hydrophobic interaction with W2856.48, F2816.44, I1143.40, T1113.37 and C1103.36. With respect to hydrophilic contacts, protonated N atom has salt bridge with D1063.32, and this cation also interacted with electron-rich aromatic residue F3127.39. Piperazine ring was also involved in hydrophobic interaction with V1073.33, V1855.39 and F2886.51.
In this binding model, we noticed that the 2-hydroxypropanol linker displayed small extent of flexibility during MD simulation, as the orientation of R-hydroxy group behaves as a H bond donor (HBD) stably swing in the cavity between TM Ⅲ and TM Ⅴ, therefore, makes H-bond with D1063.32 or Y1845.38 alternatively (Fig. S6b in Supporting information). All of the above receptor-ligand interactions were stable during 200 ns MD simulation (Fig. S6c in Supporting information), and the ligand conformation and position does not show any significant fluctuation (Fig. S7 in Supporting information). In order to know the effect of this hydroxy group, we also synthesized the corresponding des-hydroxy compound (deoxy-17) and the resulting inhibitory activity toward α1A-AR does not change (IC50 = 20.26 nmol/L for deoxy-17 vs. 24.82 nmol/L for 17). The docking pose of deoxy-17 within α1A was shown in Fig. 2b, in which binding position does not change regardless of the existence or absence of hydroxy group. This is to some extent consistent with previous study [19], in which des-hydroxy compounds also exhibited α1D/1A subtype selectivity although with a lesser extent compared to hydroxy compounds, therefore, this hydroxy is not a necessary pharmacophore for α1A-model.
The neighboring amide group was spatially proximal to extracellular loop 2 (ECL2) of α1A, leading to interaction with water of membrane boundary (Fig. S8 in Supporting information). In addition, the N atom of quinoline also behaves as a H bond acceptor (HBA), making interaction with Y3167.43 through water, the proposed pharmacophore features of R-17 was shown in Fig. S6d (Supporting information). We then utilized this model to dock compound 28 which exhibited the best α1A antagonistic activity (IC50 = 3.537 nmol/L). As expected, the xanthene ring with stronger conjugation system makes good pi-pi stacking interaction with W1023.28, and could accommodate the upper aromatic pocket well (Fig. 2c), which in turn supported the feasibility of this binding model of α1A.
With respect to α1B, to our surprise, deoxy-17 recovered the inhibitory activity toward α1B (IC50 = 329.8 nmol/L for deoxy-17 vs. 7501 nmol/L for 17). We speculated that the fluctuation of flexible hydroxy group allowed compound 17 deviating from the pocket of α1B. According to this hypothesis, we hence docked R-17 and deoxy-17 with α1B, respectively. As expected, deoxy-17 nicely accommodated in the α1B (Fig. 3b), whereas the conformation of R-17 completely distorted and nearly get out of this pocket, therefore easily lost salt bridge with D1253.32 (Fig. 3a). It seems hydroxy group decided the selectivity between α1A- and α1B-AR, while this was not true. When another hydroxy compound 28 with comparative IC50 value (IC50 = 237.3 nmol/L for α1B) docked with α1B, it was shown that 28 fitted well and remain interaction with key residues of α1B (Fig. 3c). All the above data suggested that the selectivity between α1A-and α1B-AR was influenced by both hydroxy group and upper aromatic moiety.
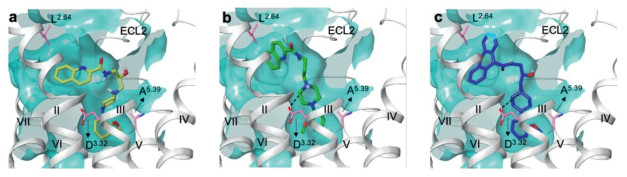
The flexible hydroxy group of R-17 lead to complete detachment from α1B, while it could stably make alternative interaction with D106 and Y184 in α1A. However, this hydroxy group of 28 was not detrimental to α1B and keep the moderate antagonistic affinity. Position 2.64 might contribute to ligand selectivity between α1A and α1B, since L2.64 in α1B allowed more space in this non-conserved cavity between TM3 and ECL2 compared to F2.64 in α1A (Fig. 2a of α1A vs. Fig. 3a of α1B). Thus, bulky xanthene ring of 28 might occupy this aromatic pocket of α1B well in spite of the existence of flexible hydroxy group. Another speculation is small size of A5.39 in α1B (Fig. 3a) expanded the orthosteric pocket so that adversely affect the key interaction between NH+ of 17 and D3.32, while V5.39 of α1A makes appropriate distance in terms of hydrophobic interaction with piperazine ring and maintained the salt bridge with D3.32 (Fig. 2a). Overall, small structural modification of ligand often makes subtype selectivity, our data supported to some extent that sequence variation within TM Ⅱ showed to be a source of subtype selectivity for α1-adrenergic ligands, however, more comprehensive study was needed.
In conclusion, we have identified a new, potent, and highly specific α1D/1A-AR antagonist, R-17, which presents a quinolinecarboxamide system and o-methyl phenylpiperazine scaffold connected by 2-hydroxy propanol linker. R-17 displays very low antagonistic affinity toward α1B, this super high selectivity of α1D/1A-ARs may contribute to the satisfied anti-BPH effect in the present study. We partly rationalize this subtype selectivity by proposing a convincing binding mode of R-17 in complex with α1A and compared with α1B binding, several key residues were proposed. Our findings may guide the optimization of phenylpiperazines to elicit a selective interaction with the desired subtype, however, more detailed and profound research should be studied in near future.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Natural Science Foundation of Guangdong Province (Nos. 2021A1515010101, 2021A1515011372, 2023A1515011895), National Natural Science Foundation of China (Nos. 21807017, 82273759, 32371529), Guangzhou Medical University Scientific Research Capacity Improvement Project (No. 02-410-2405104). We thank Professor Tomohiko Ohwada for technical support of calculation part.
Supplementary material associated with this article can be found, in the online version, at doi:
R.C. Langan, Urology 46 (2019) 223–232.
S. Madersbacher, N. Sampson, Z. Culig, Gerontology 65 (2019) 458–464. doi: 10.1159/000496289
B.M. Launer, K.T. McVary, W.A. Ricke, G.L. Lloyd, BJU Int. 127 (2021) 722–728. doi: 10.1111/bju.15286
B. Chughtai, J.C. Forde, D.D.M. Thomas, et al., Nat. Rev. Dis. Primer 2 (2016) 16031. doi: 10.1038/nrdp.2016.31
D.T. Price, D.A. Schwinn, J.W. Lomasney, et al., J. Urol. 150 (1993) 546–551. doi: 10.1016/s0022-5347(17)35544-1
M. Yoshida, J. Kudoh, Y. Homma, et al., Clin. Interv. Aging 6 (2011) 161–172.
J.R. Docherty, Eur. J. Pharmacol. 855 (2019) 305–320. doi: 10.1016/j.ejphar.2019.04.047
G. Chiu, P.J. Connolly, S.A. Middleton, et al., Expert Opin. Ther. Pat. 18 (2008) 1351–1360. doi: 10.1517/13543770802571659
D. Guo, J. Li, H. Lin, et al., J. Med. Chem. 59 (2016) 9489–9502. doi: 10.1021/acs.jmedchem.6b01217
F. Zhao, J. Li, Y. Chen, et al., J. Med. Chem. 59 (2016) 3826–3839. doi: 10.1021/acs.jmedchem.5b02023
M. Pallavicini, R. Budriesi, L. Fumagalli, et al., J. Med. Chem. 49 (2006) 7140–7149. doi: 10.1021/jm060358r
R. Takei, I. Ikegaki, K. Shibata, et al., Jpn. J. Pharmacol. 79 (1999) 447–454. doi: 10.1254/jjp.79.447
J.J. Huang, Y. Cai, Y.Z. Yi, et al., Eur. J. Pharmacol. 791 (2016) 473–481. doi: 10.1016/j.ejphar.2016.09.009
J. Huang, F. He, M. Huang, Eur. J. Med. Chem. 96 (2015) 83–91. doi: 10.1016/j.ejmech.2015.04.005
J.J. Huang, Z.H. Zhang, F. He, et al., Bioorg. Med. Chem. Lett. 28 (2018) 547–551. doi: 10.1016/j.bmcl.2018.01.068
Q. Xiao, Q.M. Liu, R.C. Jiang, et al., Front. Pharmacol. 11 (2021) 594038. doi: 10.3389/fphar.2020.594038
Q. Liu, Q. Xiao, X. Zhu, et al., Eur. J. Pharmacol. 870 (2020) 172817. doi: 10.1016/j.ejphar.2019.172817
G.H. Kuo, C. Prouty, W.V. Murray, et al., J. Med. Chem. 43 (2000) 2183–2195. doi: 10.1021/jm9905918
M. Deluigi, L. Morstein, M. Schuster, et al., Nat. Commun. 13 (2022) 382. doi: 10.1038/s41467-021-27911-3
H. Fang, M.Y. Li, L. Xia, Chin. Chem. Lett. 18 (2007) 41–44.
E.S. Stoddart, S. Senadheera, I.J.A. MacDougall, et al., PLoS One 6 (2011) e19695. doi: 10.1371/journal.pone.0019695
L. Fumagalli, M. Pallavicini, R. Budriesi, et al., J. Med. Chem. 56 (2013) 6402–6412. doi: 10.1021/jm400867d
X. Zhao, M. Yuan, B. Huang, et al., J. Mol. Graph. Model. 29 (2010) 126–136. doi: 10.1016/j.jmgm.2010.05.002
A.J. Noble, R. Chess-Williams, C. Couldwell, et al., Br. J. Pharmacol. 120 (1997) 231–238. doi: 10.1038/sj.bjp.0700907
C. Hosoda, A. Tanoue, M. Shibano, et al., Br. J. Pharmacol. 146 (2005) 456–466. doi: 10.1038/sj.bjp.0706325
S.W. Seto, S. Bexis, P.A. McCormick, et al., Eur. J. Pharmacol. 644 (2010) 113–119. doi: 10.1016/j.ejphar.2010.06.035
M.T. Quiles, M.A. Arbós, A. Fraga, et al., Prostate 70 (2010) 1044–1053. doi: 10.1002/pros.21138
N.G. Bahey, E.A.E. El-Drieny, J. Microsc. Ultrastruct. 3 (2015) 75–81. doi: 10.1016/j.jmau.2015.01.002
R. Barbaro, L. Betti, M. Botta, et al., J. Med. Chem. 44 (2001) 2118–2132. doi: 10.1021/jm010821u

Scheme 2 (1) Synthesis of target compounds. Reagents and conditions: (a) reflux, 12 h, 86.7%; (b) i-PrOH, o-methoxy phenyl piperazine, reflux, 12 h, 89.9%; (c) NH2NH2.H2O, EtOH, r.t., 89.05%; (d) HATU, DIPEA, CH2Cl2, N2, r.t., 12 h, 51.1%–87.5%. (2) Resolution condition: CHIRALCEL OZ-H, 250 mm × 10 mm, 5 µm, Semi-Prep, MeOH: DEA = 1000:1 (v/v), UV 254 nm, 35 ℃.

Figure 1 In vivo BPH model investigation of R-17 and S-17. (a) Histological changes of rat prostate tissue in various groups, including sham operated, model control, NAF (10.0 mg/kg), R-17 LOW (2.0 mg/kg), R-17 HIG (5.0 mg/kg), S-17 LOW (2.0 mg/kg), S-17 HIG (5.0 mg/kg); and quantitative analysis of epithelial thickness (µmol/L) in each group. (b) α-SMA expression of rat prostate tissue in each group and quantitative analysis of α-SMA positive cell layer thickness (µmol/L). (c) PCNA expression in the prostate tissue determined using Western blot. (d) Expression of α1A-, α1B- and α1D-adrenoceptors and (e) AR of rat prostate tissue. Columns, mean values; error bars, S.E.M. (n = 5). Significance was determined using one-way ANOVA coupled with Tukey's multiple comparisons test. #P < 0.05 compared with the sham operated group. P < 0.05, **P < 0.01, ***P < 0.001 compared with BPH model group. △△△P < 0.001. NS, no significance, P > 0.05.

Figure 2 R-17 binding pocket (a) with human α1A and ligand docking of deoxy-17 (b) and 28 (c). Ligand was shown as thick tube in yellow (R-17), green (dexoy-17), blue (28), respectively; helixes were shown as ribbons in silver color, amino acids were shown as thin tube in silver color; black dash line represent H bond or salt bridge, blue dash line represent aromatics including pi-pi stacking and pi-cation interaction.

Figure 3 Ligand docking of R-17 (a), deoxy-17 (b) and 28 (c) with human α1B, respectively. Ligand was shown as thick tube in yellow (R-17), green (dexoy-17), blue (28), respectively; helixes were shown as ribbons in silver color, amino acids were shown as thin tube in pink color; black dash line represent salt bridge.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:





 下载:
下载: