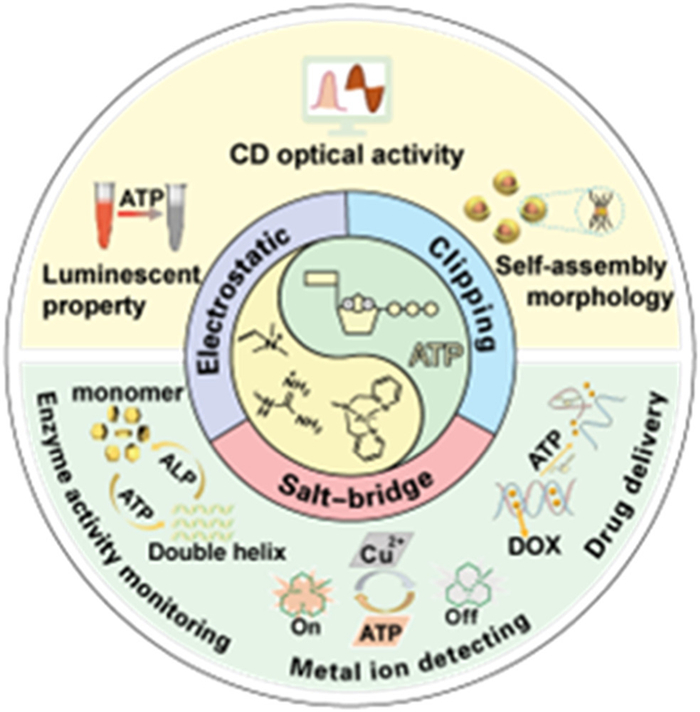
Figure 1.
Summary of ATP-induced small molecules assembly, optical properties and sensing applications.
ATP-induced supramolecular assembly based on chromophoric organic molecules and metal complexes
Zhu Shu , Xin Lei , Yeye Ai , Ke Shao , Jianliang Shen , Zhegang Huang , Yongguang Li

Phosphate derivatives, for instance, nucleoside polyphosphates, phosphatide, as well as phosphoproteins, represent vital anionic entities that assume crucial roles in cellular biology [1-4]. Among them, adenosine triphosphate (ATP) bearing three phosphate moieties with negative charges, stands as a popular exemplar of a nucleoside polyphosphate. As a ubiquitous substrate in numerous biological reactions, ATP provides the primary energy, fueling the molecular motors in living organisms within our bodies [5-8]. Its indispensability as a metabolite and critical biological signaling molecule also underscores its pivotal involvement in diverse cellular activities [9,10]. Perturbations in ATP levels have been closely linked to pathological conditions including cardiovascular diseases, cerebral ischemia, hypoglycemia, and Parkinson's disease [11-14]. ATP also assumes the role of an essential neurotransmitter [15] and exhibits a propensity for assembly [16]. It facilitates fast ligand-gated synaptic transmission at neuro-neural synapses [17], while Zn2+ appears to regulate ATP's excitatory effects on mammalian neurons [18]. ATP also acts as a substrate for signal-regulating enzymes such as adenylyl cyclase as well as kinases, playing a critical role in storing and retrieving intracellular biological information.
Recent advances highlight extensive exploitation of instant self-assembly processes in nature [19-21] involving molecules like ATP and guanosine triphosphate (GTP) with functional performances through dissipative high energy [22,23]. Nature's vast repertoire serves as a wellspring of inspiration for novel ideas, particularly the endeavor to mimic or replicate the intriguing properties of biological systems in non-living counterparts, which has garnered significant attention. The study of supramolecular self-assembly, a ubiquitous process in cellular environments and a prevalent phenomenon in nature, has witnessed widespread exploration across various disciplines. At the cellular and molecular scales, self-assembly represents an orderly arrangement of individual constituents into specific proteins or functional nanostructures, facilitated by weak noncovalent interactions in a state of thermodynamic equilibrium [24]. Supramolecular chemistry, focused on thermodynamically driven non-covalent self-assembly processes, presents a powerful tool for realizing intricate structures in artificial systems that emulate biological constructs. These endeavors not only advance our understanding of life's origins in nature but also enable the development of multiplex structures with bio-inspired features [25-32]. Consequently, ATP-depletion-mediated self-assembly has garnered significant interest in supramolecular chemistry. By employing ATP as a co-assembling component, multi-responsive systems can be designed, and ATP can serve as a trigger in self-assembly processes by facilitating structural transitions. ATP, with its amphipathic chemical structure, primarily comprises a hydrophobic and planar aromatic pyrimidine base linked to a moderately polar ribose unit, which, in turn, connects to a highly hydrophilic triphosphate moiety bearing four ionizable units. Under physiological conditions, ATP is generally regarded as a hydrotrope [33,34], facilitating the solubilization of hydrophobic components through self-association. Additionally, the four ionizable groups demonstrate pH-dependent degrees of ionization.
The distinctive molecular characteristics of ATP provide excellent design principles for self-assemblies when utilizing ATP as a component. Various weak non-covalent interactions, including electrostatic interactions, hydrogen bonding, π–π interactions, and host-guest interactions, play pivotal roles in self-assembly processes. In the realm of synthetic organic supramolecular chemistry, similar to DNA/RNA aptamers, the integration of ATP provides alternative strategies for construction of complexity building blocks and co-assembly in diverse self-assembly systems. Moreover, ATP-fueled systems feature prominently in the emerging field of non-equilibrium self-assembly [19,20,23,35-38]. However, in most cases, the ionic properties of ATPs, featuring up to ATP4− ions, preferentially form intense electrostatic assemblies with positively charged polyelectrolytes or receptors [39,40].
Optical signal is highly sensitive, non-invasive and non-contact remote control, which can be directly observed by naked eyes as well as optical instruments. Once chromophores are introduced to the various ATP systems, the self-assembly processes can be conveniently monitored, offering an approach to investigate the assembly mechanism [41-46]. Based on the self-assembly of ATP and chromophores induced optics changes, they are explored as biosensors including monitoring enzyme activities [47-49] and ATP-responsive drug carries [50-57]. On the other hand, metal ions such as Mg(Ⅱ), Ca(Ⅱ), Zn(Ⅱ), Fe(Ⅱ), Fe(Ⅲ), Cu(Ⅰ), and Cu(Ⅱ) also play irreplaceable roles in the biological systems of living organisms. Pathological disorders and neurodegenerative diseases are usually caused if the metal ions are out of the balance in the biological system [58-62]. The supramolecular assembly of ATP containing free organic ligands can cause drastically optical property changes which can be visualized and detected by instruments, once coordinated with metal ions [63-66]. Importantly, it provides a powerful strategy to better understand the biochemical processes through real-time monitoring the spectroscopic changes. Combined with the anti-cancer drugs, it can be utilized to monitor the release of drug vectors for cancer treatment due to the remarkably high level of ATP expression in cancer cells.
So far, ATP-driven assembly systems and materials, especially their equilibrium and non-equilibrium self-assembly from mechanisms, morphologies, architectures to application in biology, chemistry and materials have been reviewed [67-74]. In this concise review, we mainly focus on ATP-induced supramolecular assembly with chromophoric organic molecules and metal complexes (Fig. 1). Firstly, we introduce different ATP recognition motifs, primarily centered around cationic receptor groups. We also summarize the photophysical properties and optical activities arising from ATP-induced self-assembly, including significant spectral changes and induced circular dichroism. Furthermore, we discuss the diverse architectures exhibiting various morphologies under ATP regulation. The precise modulation of self-assembly morphologies, encompassing desired geometry, dimension, and shape-modifiable behavior, can be achieved by controlling ATP stimulation levels. Finally, we delve into the application areas of ATP, specifically its role in detecting metal ions, monitoring enzyme activity and drug delivery as well as the existing challenges and future prospects.
Nucleotides are essential biological phosphate molecules that serve as the building blocks of DNA and RNA. They consist of one or more phosphate groups connected by phosphoanhydride bonds, which release significant amounts of energy during hydrolyzed process. Among nucleotides, ATP shows a key role in cellular energy transfer, acting as the molecular currency for hundreds of cellular reactions [75]. ATP can be considered to consist of the purine base adenine, the sugar ribose, and a triphosphate group linked by phosphodiester bonds [76]. The phosphate moiety of ATP facilitates multivalent electrostatic interactions, the adenine base contributes hydrophobic and hydrogen bond interactions, and the ribose group also participates in hydrogen bonding. The unique structure of ATP, with its various non-covalent binding modes, makes it an important natural supramolecular synthesizer and has been extensively studied in ATP-induced self-assembly systems. Furthermore, ATP bearing chiral d-ribose groups is inclined to induce supramolecular chirality [77]. Various receptors exhibit diverse binding modes with ATP, particularly within the cationic receptor group (Fig. 2). For instance, ammonium and guanidine groups bind to ATP primarily through electrostatic interactions, with guanidine groups also forming salt bridges. Metal complexes, such as zinc cations, interact with ATP through a clipping mechanism. The details and pictures reported by some research groups are shown in Supporting information (Section 2.1).
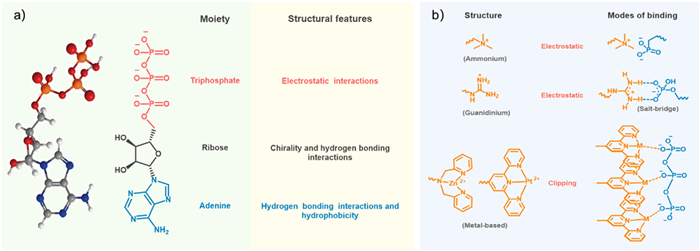
The design of ATP-induced self-assembly involves the incorporation of receptor units into the chromophore of fluorescent signals, enabling the binding of ATP through diverse mechanisms. The ammonium group serves as an efficient mediator for ATP binding, primarily through electrostatic interactions. On the other hand, the guanidinium group forms a "salt bridge" interaction, which encompasses cumulative hydrogen bonding, electrostatic interactions, together with proton transfer. This unique property allows for the incorporation of guanidine receptors into various synthetic monomers, expanding their applicability. Lastly, metal groups facilitate ATP binding through a clipping mechanism. These distinct approaches showcase the versatility and potential of receptor-based strategies in ATP-induced self-assembly.
ATP, as a fundamental building block to construct the optical-active superstructures, is deemed as a standard reference in the related optical materials field [78,79]. The electrostatic interaction between the four negatively charged triphosphate functional groups on ATP and positively charged receptors facilitates molecular proximity, leading to not only supramolecular assembly but also notable optical changes such as UV–visible (UV–vis) absorption and near-infrared (NIR) emission, as well as distinct optical activities.
Phosphorescent organoplatinum(Ⅱ) complexes, for instance, are renowned for their uniquely square-flat configuration and propensity for the formation of non-covalent metal-metal interactions, through which molecular aggregation are usually induced resulting in variable luminescent properties [80-89]. The ATP-induced supramolecular assembly of cationic alkynylplatinum(Ⅱ) terpyridine complex (10) has been investigated by Yam's research group [90]. The complex 10 is emissive at visible regions. When 10 was introduced to an aqueous buffer solution containing ATP at room temperature, the enhanced emission intensity in the low-energy NIR region at 768 nm was observed accompanying with the appearance of a broad absorbing shoulder at 510 nm, due to the formation of metal-metal and π-π packing interactions. Obviously, the NIR emission is originated from the metal-metal-to-ligand charge transfer (3MMLCT). The emission intensity was also found to increase gradually upon increasing the ATP concentration (Fig. 3a).
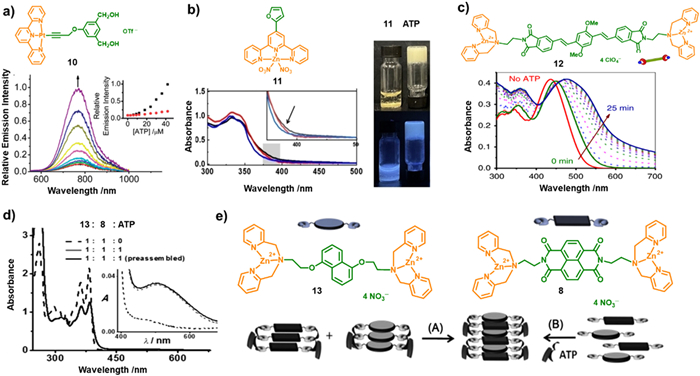
Zn(Ⅱ)-coordinated complexes present another typical species to investigate their optical behaviors in the presence of ATP. Liu et al. synthesized a series of terpyridine-Zn(Ⅱ) complexes derived with furan, thiophene, and pyrrole substitutes [91]. The 11 can be used to detect ATP from adenosine diphosphate (ADP), adenosine monophosphate (AMP), cytidine triphosphate (CTP), GTP, and uridine triphosphate (UTP) with the formation of gels, accompanied by blue fluorescence emission (Fig. 3b). In aqueous media, absorption bands at approximately 330 and 345 nm were appeared for 11, originating from π→π* ligand center (LC) transition of the terpyridine moiety. Upon addition of ATP (0.25 equiv.), the observation of a slight increase in intensity at 368 nm in the 11/ATP system suggests the formation of co-assemblies.
Acceptor-donor-acceptor (A-D-A) molecules are also commonly explored to construct supramolecular architectures with fascinating photophysical properties. The A-D-A complex 12, comprising the π-conjugated oligo(p-phenylacetylene) and DPA-Zn(Ⅱ) phosphate moieties as donor and acceptor, respectively, was designed and prepared by George and coworkers [21]. Owing to its A-D-A structure, the complex exhibited a dormant state by sliding packing model, however, the reassembly process can be triggered after the addition of ATP. UV–vis absorption spectroscopy was employed to investigate the ATP-induced self-assembly process (Fig. 3c). 12 can well dissolved in CH3CN, while in HEPES/CH3CN mixtures, short aggregates were formed. Intriguingly, upon introduction of ATP, the reassembly of 12 was induced with a time-dependent gradual red-shift absorption band from 448 nm to 478 nm, as depicted in Fig. 3c. The UV–vis spectra of 12 suggests a nucleation-growth self-assembly mechanism was adopted upon the addition of ATP. Compared with molecular receptors possessing monodentate site, ATP can induce a redshift in the absorption spectrum when combined with multi-components. The same group further reported the ATP-induced assembly of 8 and 1,5-dialkoxynaphthalene (DAN) (13) bearing DPA functional pendants (Figs. 3d and e) [92]. After addition of ATP to 13, a gradual redshift of the UV–vis absorption band was observed with decreasing intensity. Meanwhile, the intensity of the monomer emission band at 342 nm was gradually decreased, accompanying with the appearance of a new emission band at 472 nm. The prominent bathochromic emission band indicate a preassembly of 13 induced by the stacking of the chromophoric pendants in the ground state due to the absence of vibrational characteristic of the excimer emission. Further investigation of the heterodimerization of 13 and 8 was carried out to construct the charge transfer (CT) assemblies. After addition of 1 equiv. ATP to the preassembled 13 and 8, the appearance of absorption band at 510 nm suggests the formation of complexes between the D-A pairs of 8 and 13 with a ground-state CT characteristic. The in situ assembly of 13 and 8 monomers and ATP was also observed the ground state CT absorption band within a few seconds (Fig. 3d). The spectroscopic investigations reveal the ATP clippers played a key factor in facilitating appropriate chromophore preorganization from excimer to ground CT state formation.
Further expanded the π-conjugate donor moiety, perylene bisimide (PBI) chromophore (14) modified with a terminal DPA-Zn(Ⅱ) receptor motif was synthesize (Fig. 4a) [93]. 14 is in the monomolecular state in CH3CN due to its good solubility, while the introduction of water induced interchromophoric binding facilitated by hydrophobic-hydrophobic and π-π stacking interactions. To monitor the spectral changes during ATP titration, the researchers examined the UV–vis absorption spectra. They observed a gradually decrease in absorbance as well as the broadening of the absorption band until no significant change in λmax upon addition of ATP (0.5 equiv.). However, the peak maxima underwent a redshift, from 499 and 535 nm to 514 and 564 nm, respectively, finally reaching saturation at 0.8 equiv. ATP exceeding. Additionally, as the ATP concentration increased from 0.5 equiv. to 1 equiv., the emission intensity around 550 nm of H1-aggregates was gradually decreased along with the increasing emission intensity around 675 nm of H2-aggregates, accompanied by a gradual decrease in H1-aggregate emission (Fig. 4b). These obtained spectra indicate that the increased concentration of ATP induced conformations transformation from H1- to H2-state. The results provide valuable insights into the impact of ATP on the conformations and behavior of the PDPA assembly.
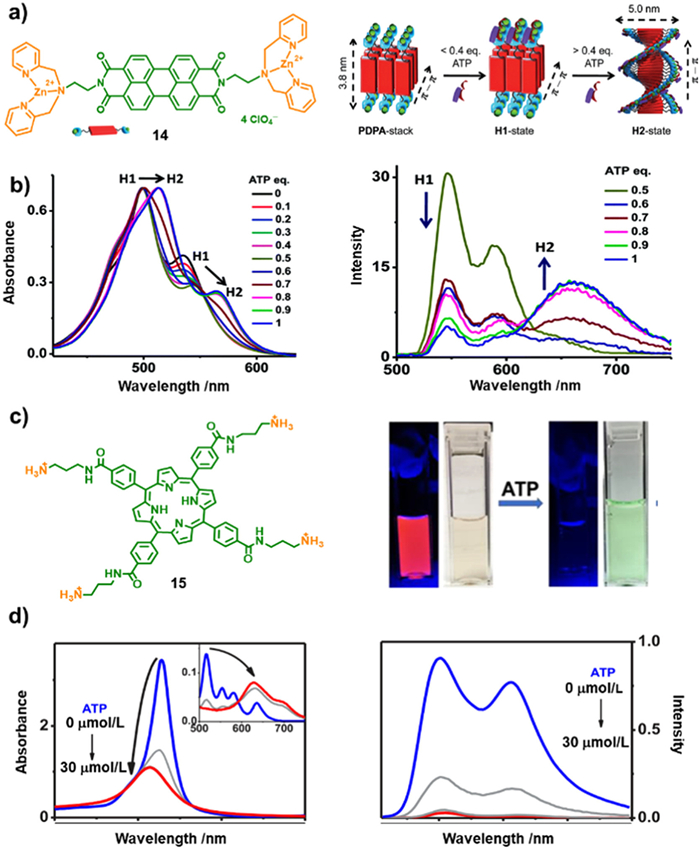
The anionic polyphosphate was also found to accelerate the aggregation of conjugated polymers featuring cationic ammonium siding groups. This aggregation behavior resulted in a redshift and quenching of the polymer fluorescence [94,95]. Based on this observation, Schanze et al. investigated the assembly behaviors of a porphyrin derivative with ammonium cations (15) induced by ATP [96]. The self-assembly process occurred under the cooperation of electrostatic and noncovalent interactions between 15 and polyphosphate negative ions. To characterize the formation of porphyrin aggregates, UV–vis absorption and emission spectroscopies were employed. These techniques are well-suited for determining the aggregate mechanism [97-102]. The addition of ATP induced self-quenching of porphyrin fluorescence resulting in a reduction in the Soret transition extinction (Figs. 4c and d). The naked color drastic changing from colorless to light green provides a visual method of ATP sensing capabilities (Fig. 4c). The blue-shift of the Soret band indicates the formation of H-aggregate, accompanied by fluorescence quenching of the porphyrin molecules (Fig. 4d). The absorption and fluorescence data suggested the presence of strong interchromophore interactions within the 15-ATP assembly, potentially arising from the stacking of porphyrin macrocycles to form "card-wrapped" H-aggregate structures.
ATP can also serve as a chiral gene to induce supramolecular chirality during the assembly process, primarily attributing to the presence of D-ribose groups. For instance, the cationic porphyrin 15 also exhibited supramolecular induced chiroptical properties in the presence of ATP [96]. The positive (402 nm) and negative (415 nm) circular dichroism (CD) signals with isochronic point observed at 405 nm, immediately emerged and intensely increased upon the addition of ATP, which is in line with the Soret band region in the UV–vis absorption spectra. Additionally, time-dependent CD spectra of 15-ATP showed that the CD intensity alleviated increasing with time and reached its maximum after 30 min. These observations suggest that the self-assembly of 15-ATP complexes can be attributed to the robust electrostatic interactions between the porphyrin cations and phosphate anions, yielding supramolecular CD-activity. The CD signals also elucidated that these assemblies provided an optically active environment for the porphyrin chromophore, indicative of a helical structure.
George and his colleagues demonstrated that in the ATP-induced PDPA assembly system, no monomers 14 could be found indicating its complete assemble [93]. Subsequent CD titration experiments revealed that the CD signal remained almost inactive until the introduction of ATP (0.5 equiv.) with the formation of H1 state, but the signal abruptly increased upon further adding of ATP and reached its maximum till the addition of one equiv. of ATP with the aggregation transformation from H1 to H2 state. The CD spectra exhibited a positive (518 nm) and negative (480 nm) maxima, with a zero crossover (507 nm). The CD spectra were also in line with the PBI UV–vis absorption spectra (Fig. 5a). This also suggested the right-handed supramolecular helical structures of the PBI chromophore was formed in the H2 state (Fig. 5b).
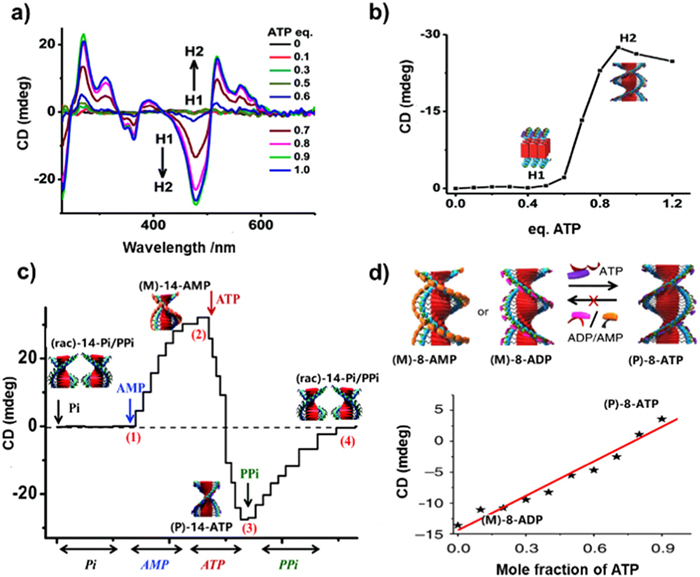
Similar to ATP, the phosphate analogues such as Pi, AMP, pyrophosphoric acid (PPi), were also investigated to study their influence on the chirality transfer in supramolecular systems. George's group demonstrated that a complete cycle of helix mutation could be realized through the refined modulation of preassembled architectures via continuously exchanged negative charged phosphate analogues [103]. Initially, the racemic assembly (rac)-14-Pi was formed by π-stacking of molecules 14 and electrostatic interaction among Pi and 14, due to the inorganic monophosphate Pi[(PO4)3−]. Subsequently, AMP was gradually added to the above system leading to the formation of (M)-14-AMP, which was confirmed by the emergence of a negative bisignate CD signals. Upon further addition of ATP, the CD signal underwent a reversal with the emergence of a positive bisignate signal instead of the negative signal, accompanying with an isochromatic point with a zero crossover at 423 nm and yielding a (P)-14-ATP. While, as the diphosphate PPi was introduced to the (P)-14-ATP stack, the intensity of the CD spectra was gradual decrease, eventually leading to complete CD-silence at high PPi concentrations. This transition indicated the transformation from (P)-14-ATP to (rac)-14-PPi stacks (Fig. 5c). This dynamic helical inversion among (rac)-14-Pi/PPi, (M)-14-AMP and (P)-14-ATP helix indicating the counterions exchange strategy to refine the chirality of the supramolecular is efficient.
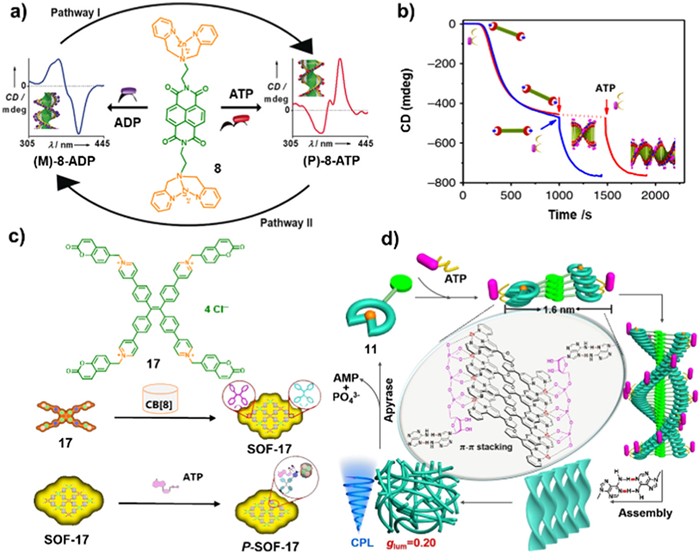
Based on the counterions exchange induced chirality transfer and amplification concept, they further demonstrated the tunable and dynamic helicity switching of chiral amphiphilic peptides (APs). Another examples are the chromophores NDI derivatives 8 and 9 could be induced possessing supramolecular helical assembly upon addition of ATP [104]. The compound 8 exhibited an opposite chirality when bound to ADP or ATP. The presence of AMP or ADP induced a left-handed (M-helicity) conformation in the NDPA assembly, according to the negative bisignated CD spectrum. However, the addition of ATP induced the chiral inversion, resulting in a right-handed (P-helicity) arrangement, according to the positive bisignated CD spectra. The different adenosine phosphates induced contrasting chirality in the NDI assemblies, prompting the researchers to investigate dynamic helical inversion through binding competition among multivalent phosphate derivatives. The researchers conducted titration experiments using compound 9 in the presence of ADP (0.5 equiv.). To the above supramolecular systems, ATP were gradually added. As the content of ATP reached to 0.5 equiv., a positive bisignated CD signal emerged, in accordance with the CD signal obtained from 9-ATP directly assemblies. Similar chirality reversal experiment was also found of compound 8 [105]. The M-helicity species of 8-AMP and 8-ADP were formed with intense CD signals. The CD intensity of both (M)-8-AMP and (M)-8-ADP was increased upon successive addition of small amounts of ATP (Fig. 5d). CD signal inversion occurred through progressive increments in intensity during addition of each portion of ATP. Moreover, the negative to positive signals transition was indicative of a stereoscopic mutation, further suggesting (P)-8-ATP aggregates were formed through binding competition of ATP with AMP or ADP.
Since the ATP is found in organism to provide energy, the biologically inspired synthetic supramolecular systems have also explored which can be driven by controllable chemically fuels to achieve transient helical conformational transition, nonequilibrium dynamics, tunable stereo-mutation rates, and transient conformational lifetimes [23]. Two "tandem enzymes", hexokinase (HK) and creatine phosphokinase (CPK), were employed to catalyze ATP formation and decomposition to regulate the stereoscopic mutational dynamics between (M)-8-ADP and (P)-8-ATP. Upon the addition of ATP to the solution of 8 and HK, a positive Cotton effect was immediately formed, indicating the conformation formation of (P)-8-ATP. Subsequently, the transformation from (P)-8-ATP to (M)-8-ADP was triggered by HK due to the consumption of ATP and generation of ADP and glucose-6-phosphate and the process was monitored by time-dependent CD spectra. The transient conformation persisted for several hours, after which the CD signal gradually reversed and exhibited a growing negative bisignated CD signal, corresponding to a P-M stereo-mutation. The catalytic reaction was further confirmed by 31P NMR experiment. Dynamic light scattering (DLS) measurement showed no obvious scattering size changes during the reaction process as well as the initial and final states, demonstrating the conformational change from (P)-8-ATP to (M)-8-ADP without obviously disassembly of the aggregates of 8. Introduction of the sacrificial agent of phosphocreatine, (P)-8-ATP were also able to converted to (M)-8-ADP with the reversal Cotton effect (Fig. 6a). The investigation provides a new way to develop chirotechnological and biomimetic applications.
Rational designed CT complexes were also found to show remarkable amplified chiroptics [92]. Different from the formation of excimers from pure compounds 13 or 8, mixed cofacial CT complexity were formed after mixing 13 and 8 with a molar ratio of 1:1. The strongly coupled bisignated CD signal of the mixtures of chromophores 8 and 13 was observed after combining with ATP through electrostatic interaction (1 equiv.). This CT complexity exhibited distinct chiroptical spectroscopies compared to these of individually organized NDI and dipyricloethylenediamine chromophores. A broadening Cotton effect band of 350 nm was observed, accompanied by the observation of 365–332 nm zero-crossing, indicating the formation of chiral co-organized CT complexity. Furthermore, a monosignated Cotton effect band with a maximum at 500 nm appeared, in line with the absorption region originating from the CT transition, providing evidence for the presence of a hybrid CT component with strong exciton coupling and helical organization [22]. Besides, π-conjugated oligo(p-phenylacetylene) chromophore with phosphate receptors 12 was designed to study temporal supramolecular polymerization [21]. The A-D-A molecule 12 could be assembled into small aggregates in MeCN/HEPES (1:9; v/v). Upon the addition of ATP, the self-assembly process was triggered via a nucleation elongation mechanism with appearance of a positive bisignated CD signal (Fig. 6b).
Besides CD properties, circularly polarized luminescence (CPL) is another important characteristic to estimate the materials performance. For instance, a supramolecular organic framework (SOF) prepared from 17 and CB[8] was designed by Cao et al. (Fig. 6c) [106]. The molecular 17 possessed TPE and coumarin as the chromophore and polymerization functional moieties, respectively. The dimer of coumarin was formed by adopting a head-to-tail packing mode in the CB[8] cavity by the host and guest complexation. Hence, SOF-17 was obtained via homodimerization between the coumarin moieties in 17 and CB[8]. The chirality was induced by addition of ATP by electrostatic interactions between ATP and the methylated pyridine. As the ATP reached 0.4 equiv., a significantly enhanced CD signal of a negative Cotton effect from 323 nm to 406 nm was detected. This enhancement was attributed to the formation of chiral P-SOF-17, as a consequence of the stronger binding affinity between SOF-17 and ATP. The high binding interaction limited the corotating conformation of the TPE unit within the SOF structure, enhancing the chirality transfer and aggregation-induced emission behaviors. The ATP induced P-SOF-17 exhibited right-handed supramolecular CPL property with a luminescence dissymmetry factor (glum) of −10−4. These findings provide compelling evidence that the chiral properties of ATP can be transferred to the SOF-17, leading to the formation of P-SOF-17 or M-SOF-17 with dynamic CPL characteristic. Other adenosine derivatives, peptides as well as protein could be also interacted with SOF-17 with high-performance CPL behaviors.
Liu et al. developed a serials of metal coordinated complexes with specific identification of ATP with a remarkable glum = 0.2 from other nucleotides derivatives including ADP, AMP, UTP, GTP, CTP and so on [91]. Upon addition of ATP, the furan substituted terpyridine-Zn(Ⅱ) complex 11 exhibited Cotton effect with a positive signal at 368 nm in the CD spectrum and a distinct positive CPL band at 460 nm indicating the presence of chirality information in the excited state. The researchers demonstrated that helical fibers was formed due to the following factors, including the coordination between the Zn(Ⅱ)-terpyridinium groups and the phosphate anions of ATP, the π-π stacking interactions among the conjugated systems of 11, and the intermolecular hydrogen bonding interactions. Subsequently, the helical fibers intertwining together with the formation of hydrogel, resulting in the remarkable CPL emission (Fig. 6d). So far, this is the first example to utilize CPL to detect ATP from the analogues.
Overall, the interaction between ATP and various rational designed molecular systems can induce supramolecular assembly, alter optical properties, and regulate conformation transformations, providing a strategy for the development of functional molecular materials and sensors.
The ATP-mediated supramolecular assembly of different functional molecules not only brought distinct optical and chiroptical properties, but also showed diverse morphologies, which might have effect on the optical performances. Some relevant scientific research results are described in Supporting information (Section 2.4). Study from the activity of living organisms, the dynamic assembly of the artificial synthesized organic molecules, metal coordinated complexes as well as copolymers can be achieved driven by ATP fuels. The concentration of ATP plays a crucial role in controlling the assembly morphology, while the presence of ATP hydrolase realizes the reversibility of the architectures. The research about the bio-responsive materials will be beneficial to mimic the biological activities.
It is well-known that metals such as Mg, Ca, Zn, Fe, and Cu play crucial roles in the biological systems of living organisms [107,108]. For instance, these metals are essential for various biological processes, including enzyme catalytic activity and the maintenance of structural stability in chromosomes [109-111]. However, both deficiency and excess of these metals can lead to variously pathological disorders, such as gastrointestinal disturbances, liver or kidney damage, and neurodegenerative diseases [112]. Therefore, the detection of metals is of great importance in biomedical and environmental science. It is imperative to develop probes that possess high selectivity and sensitivity to detect metal ions. Hence, in this section, we present some simple approaches for metal ion detection based on the supramolecular complex formation between cationic polymers and ATP.
In 2013, Wu et al. reported a visualized colorimetric probe to probe Cu(Ⅱ) in water. They utilized a cationic polythiophene derivative and ATP as chromophore and fuels, respectively, to construct the probe. The probe exhibited a low detection limit of Cu(Ⅱ) (0.05 mmol/L) [63]. The interaction between Cu(Ⅱ) and ATP propelled the dissociation of the complexity between polythiophene derivative and ATP. This led to a conformational conversion of the polymer chain, resulting in a visible color change in the probing system from pink-red to orange with variable Cu(Ⅱ) (Fig. 7a). Recently, a luminescence sensor, named Mito-A (18), was designed and synthesized as shown in Fig. 7b [65]. The luminescence of Mito-A was quenching upon the coordination between 18 and Cu(Ⅱ), resulting in the luminescence turn-off. Upon addition of ATP the fluorescence was turn-on immediately due to the competed binding interaction with Mito-A.
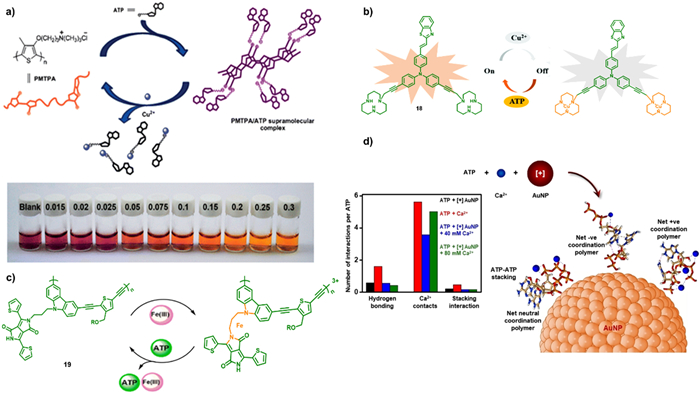
Besides the phosphorescence metal coordinated complexes, the polymers are also explored as luminescent sensor. For example, diketopyrrolopyrrole pendant poly(carbazole-alt-thiophene) (19), a π-conjugated luminescent polymer was prepared via Sonogashira coupling polymerization using Pd(Ⅱ) as catalyst [113]. This emissive π-conjugated macromolecular probe exhibited selectivity towards Fe(Ⅲ), resulting in the quenching of the orange emission. However, upon introducing ATP to the metallopolymer, a significant enhancement in emission intensity is observed as illustrated in Fig. 7c. This emission enhancement of the polymer is due to the coordination between ATP and Fe(Ⅲ) through electrostatic interactions.
Ca(Ⅱ) was also probed by AuNPs in the presence of ATP [64]. These cationic AuNPs overcome the electrostatic repulsion between ATP molecules, leading to the precipitation of AuNP-ATP aggregates. However, upon the addition of Ca(Ⅱ) to the system, the charges are screened and coordination interaction occurred between ATP and Ca(Ⅱ). The coordination, in turn, regulated the strength of electrostatic attraction between negatively charged ATP and positive charged AuNPs as depicted in Fig. 7d.
The relative studies provide valuable insights into the detection of metal ions using supramolecular complexes of cationic polymers and ATP. The developed colorimetric and fluorescent probes exhibit high selectivity and sensitivity for specific metal ions, enabling their detection in various biological and environmental contexts. These findings contribute to the advancement of metal ion sensing methodologies and significant potential for diverse applications.
ATP, well-known as a nucleoside polyphosphate, is widely recognized as a critical energy source for various metabolic pathways. It undergoes hydrolysis catalyzed by ATPase, leading to the formation of ADP and Pi, thus releasing free energy. ATP also serves as a phosphate donor in kinase-catalyzed phosphorylated reaction. Kinases, as phosphorylases, utilize ATP to transfer phosphate groups to specific substrates [114]. Conversely, alkaline phosphatase (ALP), a membrane-bound enzyme present in almost all living organisms' tissues, exhibits broad substrate specificity, catalyzing the hydrolysis or transphosphorylation of diverse phosphate compounds in vitro. ALP is known for its non-selective hydrolysis of phosphoanhydride bonds, decomposing ATP into inorganic Pi and organic adenosine. Several approaches are explored to study the kinetics of the crucial enzymes, including radioisotope-based assays, modified substrates, and laborious sampling and analysis procedures. However, there is still necessary to develop more convenient preparation and detection processes. This section focuses on several detection probes that not only determine target substrates but also facilitate the convenient monitoring of enzyme kinetics involved in these catalytic transformations.
The terpyridine platinum(Ⅱ) complex 10 with hydroxyl ligand was demonstrated to be shown self-assembly properties in aqueous solution, which can be governed by the polyanionic nature of ATP and phosphopeptide [90]. Hence, the self-assembly behavior with drastic spectroscopic changes could be designed as a real-time probe for monitoring the enzymatic activities of ATPase. The experimental results confirmed that ATP was decomposed into ADP and Pi under the catalyst of ATPase. The aggregates of 10 and ATP were disassembly with drastic apparent color and emission changes (Fig. 8a). Schanze et al., in 2019, developed a reversible double-helix assembly system to investigate the enzymatic activity monitoring [96]. Their work focused on amino cationic porphyrin compound 15. Complexation with ATP, double helical superstructure was formed, which can be disassembly into monomers as ALP was introduced to catalyze ATP hydrolysis. Subsequently addition of ATP can trigger the reassembly of the double helical nanostructures. Meanwhile, the NIR luminescence of 15 switched off induced by the ATP-induced aggregation accompanying with distinct color changes from light yellow to green. The emission and naked eye colors can be mediated by ATP and ALP, thus provide a visualized sensor for ATP and ALP detection (Fig. 8b).
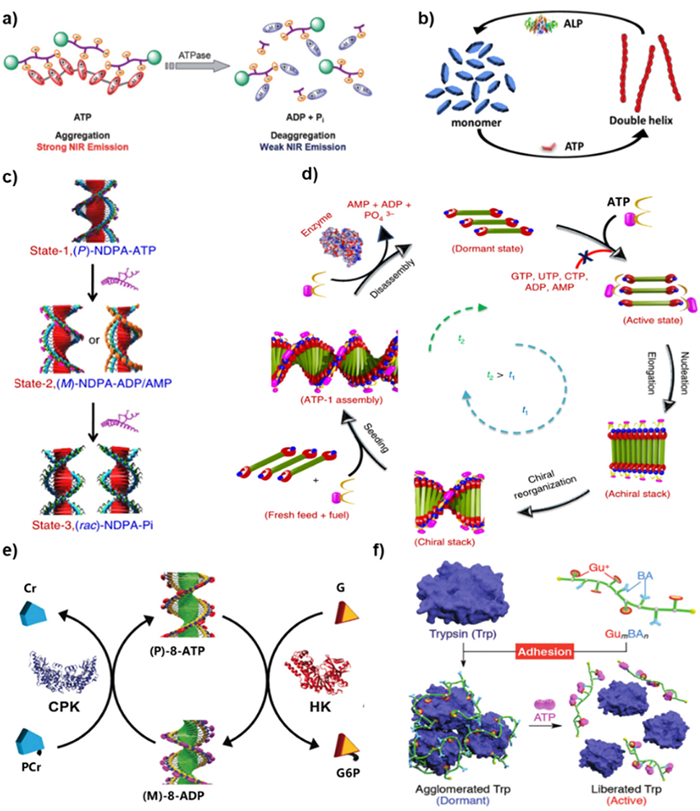
Liu et al. employed apyrase derived from potatoes ATPase to investigate the in situ hydrolysis of 11/ATP through the time-dependent CPL changes [91]. The experimental setup involved the addition of apyrase to the 11/ATP assembly, with the CPL signal monitored with time at 25 ℃. The CPL signal at 460 nm rapidly decreased within the initial 10 min, indicative of enzymatic hydrolysis drove ATP to be converted to ADP and AMP. The kinetics of this process demonstrated that the CPL signal gradually declined with increasing enzyme concentration. The CPL signaling was resumed upon the subsequent addition of ATP. Thus, the intensity of the CPL signal could be served as an indirect information for monitoring the enzyme activity through ATP. Calf intestinal alkaline phosphatase (CIAP) enzyme is recognized for its ability to catalyze the hydrolysis of all three forms of AP, resulting in the dissociation of adenosine and phosphates. A C3-symmetrical terpyridine Tb(Ⅲ) probe was developed to monitor the direct enzymatic reaction of ATP with CIAP via luminescence changes [115]. The spheroidal structure with enhanced luminescence was formed due to the noncovalent interaction between Tb(Ⅱ) complex and ATP. The luminescence would be quenched upon the addition of CIAP. CIAP was also employed to elucidate the response of chiral APs to tunable handedness and dynamically switchable helicity [105]. The experimental design involved a supramolecular helical assembly comprising of self-assembled NDI chromophores that were terminally modified with Zn(Ⅱ)-coordinated dipyricloethylenediamine receptor motifs (8). Notably, binding of AMP or ADP induced an opposite handedness in the helical assembly compared to that of ATP. Upon the addition of CIAP to the system, the helical handedness of the 8 stacks underwent dynamic reversal, owing to the conversion of ATP to ADP or AMP via enzymatic hydrolysis. This reversal of helicity was illustrated in Fig. 8c. The kinetics of this process exhibited a rapid spiral reversal with increasing enzyme concentration. In a recent study, a turn-on fluorescent probe based on AMP and Tb(Ⅲ) coordinated polymer has been developed. The probe was successfully used to detect fluoroquinolone residues in milk samples with the detection limits to 0.01 µmol/L [116].
In an attempt to mimic the growth and dynamics of actin, the researchers aimed to design a highly selective assembly process for ATP. They hypothesized that, by implementing time-controlled enzymatic hydrolysis of ATP to ADP, rapid assembly similar to actin could be achieved [21]. To demonstrate this phenomenon, potato apyrase was chosen as the enzyme for depolymerized investigation because of its high-performance enzymatic activity. Initially, the A-D-A compound 12 combined with ATP concentration at low enzyme concentration facilitated the dominant instantaneous self-assembly of the target structure. As the enzyme concentration increased, the hydrolysis of ATP became more rapid, resulting in a concomitant disassembly of the assembly with an accelerated rate. This relationship between enzyme concentration, ATP hydrolysis, and assembly disassembly is depicted in Fig. 8d.
ATP can serve as a valuable tool for monitoring the interaction between two enzymes, such as phosphatases and kinases, in a manner akin to a tandem system. To confirm this concept, George and colleagues chose NDPA, a derivative of naphthalene diimide, as a phosphate receptor [23]. They employed two kind of complementary phosphoryl transferase enzymes (HK and CPK) to construct the biologically inspired "tandem enzyme" approach. In addition to the ATP-hydrolyzing enzyme HK, CPK was utilized to induce biochemical oscillations in the circadian rhythm, leading to the formation of (P)-8-ATP through non-equilibrium processes (Fig. 8e). Specifically, HK facilitated the transfer of a phosphate group from ATP to glucose (G), resulting in the formation of glucose-6-phosphate (G6P) and ADP. Meanwhile, CPK transferred phosphate from the sacrificial reagent phosphocreatine (PCr) to ADP with the formation of ATP and the side product of creatine (Cr).
In a recent report, the diblock copolymer was designed to modulate enzyme activity (Fig. 8f). The ATP-responsive molecular glues, named GumBAn, possessed numerous Gu+ on the arm chains together with large quantities of BA pendants [117]. These molecular glues exhibited high affinities for ATP due to the formation of multiple Gu+/PO4− salt bridges and 1,2-diol/BA covalent bonds. Among them, Gu12BA12 was found to transiently inhibit the activity of Trypsin (Trp) through hybridization, potentially leading to the agglomeration of Trp and hindering access to its active site for enzyme substrates. Conversely, in the presence of ATP, the high affinity promoted ATP competitively binding to Trp, thereby dissociating Gu12BA12 and restoring the enzymatic activity of Trp. ATP and ADP can also be served as effective indicators of enzyme activity through competitive interactions with luminescent probes. A notable example of this approach was demonstrated by Prints and colleagues in 2015. They utilized AuNP passivated by a terminated thiols monolayer modified with a 1,4,7-triazacyclononane (TACN)·Zn(Ⅱ) head group as a fluorescent probe [118]. This probe exhibited distinct behavior depending on the presence of ATP or ADP. Specifically, the fluorescent probe remained freely dispersed in solution in the presence of ATP, while the probe was captured by the AuNP in the presence of ADP, leading to the luminescent quenching. The fluorescence signal exhibited a linear response to different ratios of ATP/ADP, enabling the monitoring of protein kinase activity by introducing an equal amount of enzyme and subsequently measuring the luminescent intensity.
Up to now, various detection probes have been developed to monitor the kinetics and activities of enzymes involved in ATP hydrolysis. These probes utilize self-assembly properties, luminescence changes, helical assembly dynamics, and competitive binding interactions with ATP and ADP. These innovative approaches provide convenient and efficient means to study enzyme activity and enable the monitoring of enzymatic transformations in a more accessible and practical manner.
Cancer is one of the hardest diseases to treat [119,120]. ATP, originating from biology, is essential during the process of physiological metabolism. Especially, intracellular ATP levels is remarkably higher in the cancer cells due to the enhancement of glycolytic metabolism [121,122]. ATP are deemed as a powerful biomarker for cancer diagnosis because of its key role during the proliferation and spread process of cancer cells [123,124]. Wang et al., for example, designed a hybridization chain reaction (HCR) amplification approach based on extracellular ATP activation to create three DNA probes, including H1-ATP aptamer duplex, Apt-trigger and hairpin H2 [125]. A quencher (BHQ1) and a fluorescein (AF488) were used to labeled the opposite side of the H2 stem. In order to precisely identify cancer cells, ATP aptamers can bind to activated ATP as recognition units. The isolated H1 then produces HCR with H2, amplifying the fluorescence signal. Furthermore, the control experiments from UTP, GTP, and CTP revealed that only ATP could cause a noticeable HCR and increased fluorescence intensity due to the particular site recognition of ATP and aptamers.
Meanwhile, drug delivery systems (DDSs) based on nanocarriers, with the advantages of good stability, high drug loading capacity, and controlled drug release, provide a promising application in anti-cancer therapy [126-129]. The primary purpose of DDSs is to kill cancer cells within the body. Researchers have created ATP-mediated nanocarrier DDSs because the incredibly high amount of ATP found in cancer cells can be utilized as an efficient internal stimulant to control intracellular drug release.
Doxorubicin hydrochloride (Dox) is a significant anthracycline-based hydrophobic anticancer medication, and its fluorescence characteristics can be utilized as a foundation for tracking Dox release levels. As a result, numerous nanocarriers such as mesoporous silica carriers [130,131], DNA nanocarriers [132,133], liposome nanocarriers [134], polymer nanocarriers [135,136], and nanoporous metal-organic framework (NMOF) carriers [137,138] are coated with Dox have been thoroughly investigated. Gu et al. developed an ATP-responsive DNA motif, protamine, and hyaluronic acid (HA) cross-linked shells to create a nanocarrier-based DDSs that can encapsulate Dox [54]. The HA shell can selectively be broken down by hyaluronidase as the nanocarriers entered the cancer cells. The exposed DNA motif aptamers attach to ATP with a high quantity in cancer cells, causing the nanocarriers to undergo conformational modification and selectively release Dox. The fluorescence intensity of released Dox was measured by incubating Dox/Duplex at various ATP concentrations (Fig. 9a). At the ATP concentration of 8.0 mmol/L, a notable recovery of Dox fluorescence was determined, indicating the vector's robust drug release capability at high ATP concentrations. They further explored hybrid nanoaggregates of two single-stranded DNA, graphene oxide (GO), and ATP aptamers cross-linked by graphene as Dox carriers [56]. The loading capacity of Dox is greatly increased by π-π stacking interaction between GO and Dox.
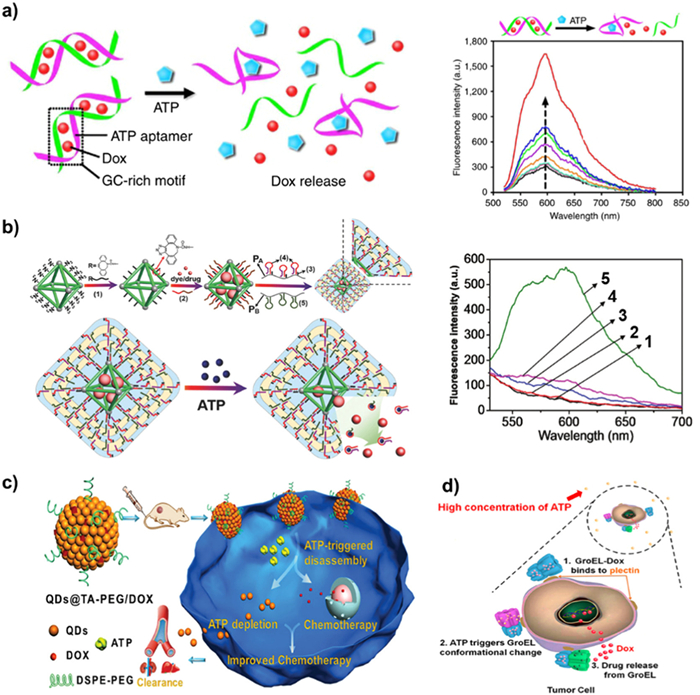
The ATP-responsive NMOF coated with hydrogel was reported by Willner and co-workers using a cross-linking technique between two polyacrylamide chains, PA and PB (Fig. 9b). Subsequently, the ATP aptamer was loaded in hairpin in Fig. 9b labeled as (4), modifying with PA chain [139]. Upon ATP binding to the ATP aptamer in cancer cells that ATP was overexpressed, breaking down the hydrogel covering and releasing either the anticancer medication Dox or Rhodamine 6G wrapped in NMOFs. The NMOFs-loaded medication had been fully released determined by the time-dependent fluorescence curve of ATP triggered Dox release. Furthermore, the rates of Dox release can be controlled by the ATP concentration. A higher degree of loaded medications will be released with the increase in ATP concentration. It was also demonstrated that the release of NMOFs and hydrogel-coated Dox exhibited selectivity because other triphosphate nucleotides, like GTP and UTP, could not cause the release of Dox. The study also showed that compared to nucleic acid-gated NMOFs, hydrogels coated NMOFs have a larger loading capacity and less leakage [57].
Chen's group used tannic acid (TA) and CdSe@ZnS quantum dots (QDs) to prepare QDs@TA phenolic nanoclusters (NCs) as nanocarriers [140]. Polyethylene glycol (PEG) was introduced to the carrier surface in order to increase their biocompatibility. The fluorescence of NCs was quenched because of the short-distance energy transfer of self-assembled QDs and the catechol group on TA. Once the QDs@TA-PEG/Dox was injected into cancer cells, due to the nature of ATP binding with metal, ATP would replace TA binding with QDs, further breakdown NCs, and release Dox (Fig. 9c). Meanwhile, the fluorescence intensity of Dox recovered after the destruction of NCs by ATP. These ATP-responsive nanocarriers with fluorescent on-off characteristics showed great benefit for DDSs and fluorescence imaging applications.
Natural proteins with double cage structures, like GroEL, can also be employed as Dox carriers. The unique structure makes GroEL a suitable delivery system for the hydrophobic medication Dox. Nie et al. discovered that elevated ATP concentrations in cancer cells caused GroEL-Dox to specifically bind to plectin, a target protein that is aberrantly expressed on tumor cell membranes [141]. Subsequently, GroEL-Dox underwent a conformational transition resulting in the conversion of a hydrophobic protein cavity into a hydrophilic one (Fig. 9d). Hydrophobic Dox was extruded and released into the nucleus, causing cancer cells to undergo apoptosis.
The chemotherapy medication pemetrexed (PMX) can also be utilized as a model anticancer agent through host-guest interaction with a water-soluble pillar[6]arene (WP6A) as carrier [142]. In order to construct a stable complex between ATP and WP6A, overexpressed ATP can substitute PMX in a competitive manner. It was discovered that the complex released PMX, which shut off the energy supply of the efflux pump and gave cancer cells a synergistic therapeutic benefit. Other studies have demonstrated that ATP can stabilize amorphous calcium carbonate and act as a source of phosphorus for coating anti-cancer medications as calcium carbonate carriers [143,144]. So far, numerous studies have shown the critical role that ATP plays in cancer treatment, particularly in the transport of medications. ATP-responsive DDSs is the primary method which break down carriers by ATP aptamers to release the drugs at tumor location and improve therapeutic effects.
ATP as a ubiquitous substrate in various biological reactions, has garnered significant attention in the field of supramolecular chemistry due to its ability to mediate self-assembly processes. Through the interaction of ATP, a series of monomeric molecular units connected by non-covalent bonds can assemble into supramolecular polymers. Under physiological conditions and at neutral pH, ATP is generally considered to be water-soluble. In ATP-induced synthetic systems, the phosphoric acid components of ATP facilitate multivalent electrostatic interactions, while the adenine bases contribute hydrophobic and hydrogen bond interactions. Additionally, the ribose groups present in ATP can participate in hydrogen bonding interactions. Moreover, ATP exhibits non-covalent chiral induction properties, allowing it to act as a chiral promoter in supramolecular assemblies due to the presence of chiral d-ribose groups. Various types of cationic receptors have been developed to bind ATP, either through electrostatic interactions or through molecular clipping.
The interaction between the positively charged receptor and the negatively charged triphosphate functional groups in ATP initiates supramolecular assembly and induces significant changes in photophysical properties. The presence of ATP leads to redshifts in the ultraviolet spectrum and enhances fluorescence. Simultaneously, ATP can promote the aggregation of luminescent groups and self-assembly, resulting in fluorescence quenching. Particularly, ATP exhibits specific binding affinity towards certain organic phosphorescent metal complexes, such as Pt(Ⅱ), which tend to engage in non-covalent metal-metal and π-π packing interactions, leading to pronounced color and emission variations upon assembly. The spectral changes observed in these systems can be utilized to monitor the extent of self-assembly. Furthermore, due to the chiral d-ribose groups present in ATP, ATP-induced self-assembly can exert control over chiral signals and even govern helical mutation cycles from racemic components to both left- and right-handed helices, ultimately transitioning into racemic stacks. This rational design, enabling unprecedented regulation of spiral mutation cycles, holds great promise for applications in switchable, enantioselective, and microtechnological systems. Several ATP-driven chiral assembly systems have also demonstrated specific recognition of ATP through CPL. The development of adaptive chiral fluorescent materials exhibiting both CD and CPL responses offers unique insights into the design of chiral functional materials and expands the potential applications of such materials.
Moreover, the morphology of self-assembled structures can be intricately controlled by precisely modulating the levels of ATP stimulation during the assembly process. This precise regulation empowers the attainment of anticipated geometries, dimensions, and shape-changing behavior. Additionally, the reversible decomposition of ATP assemblers in response to phosphatase introduces novel avenues for ion-ion interactions and enzyme-mediated transient self-assembly nanotechnology. This remarkable capability paves the way for the development of complex chiral structures and the design of active functional materials that emulate the properties of naturally occurring biomaterials. Such materials hold the potential to serve as drug release systems with controlled release capabilities tailored for biological environments.
Certain metal polymers exhibit remarkable sensitivity and selectivity for ATP over other biologically relevant anions such as ADP and AMP. This pronounced discrimination arises from the significantly stronger electrostatic interactions between the metal polymer and ATP. Notably, these metal-polymer systems manifest noticeable color changes, discernible to the naked eye under 365 nm light, making them promising candidates for ATP detection. Consequently, ATP-induced self-assembly in the context of metal ion detection offers a valuable strategy for the design and development of simple, selective, and sensitive metal-polymer-based probes to detect chemical and biologically relevant analytes. However, it is crucial to acknowledge the limitations of ATP-based supramolecular assembly. Primarily, its applicability is confined to water-soluble systems, which restricts its broader use in materials science applications. Moreover, the proposed strategy may be system-specific and not directly transferable to other synthetic systems. Additionally, in systems where added enzymes directly hydrolyze ATP, complex structural dynamics akin to those observed in the cytoskeleton might not occur. This phenomenon is influenced by the presence of excess ATP and the preferential degradation of ATP based on its solubility.
In conclusion, the field of ATP-induced synthetic supramolecular assembly has seen considerable development in recent years. It also encounters substantial challenges in the supramolecular community. By transforming ATP from a simple structural motif in supramolecular and host-guest chemistry to a responsive motif, an exciting opportunity arises for supramolecular chemists to design synthetic systems with bionic responsiveness and temporal properties. This advancement holds significant potential for creating innovative and adaptable supramolecular architectures with a wide array of practical applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (Nos. LR22B010001, LQ23B010001), the National Natural Science Foundation of China (Nos. 22201057, 21871297), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholars (No. 2019B151502051) and the Hangzhou Normal University (Nos. 2021QDL001, 2021QDL065).
Supplementary material associated with this article can be found, in the online version, at doi:
W.W. Cleland, A.C. Hengge, Chem. Rev. 106 (2006) 3252–3278. doi: 10.1021/cr050287o
M.W. Bowler, M.J. Cliff, J.P. Waltho, G.M. Blackburn, New J. Chem. 34 (2010) 784–794. doi: 10.1039/b9nj00718k
L.N. Johnson, R.J. Lewis, Chem. Rev. 101 (2001) 2209–2242. doi: 10.1021/cr000225s
M.B. Yaffe, Nat. Rev. Mol. Cell Biol. 3 (2002) 177–186. doi: 10.1038/nrm759
H.R. Xu, K. Li, S.Y. Jiao, et al., Chin. Chem. Lett. 26 (2015) 877–880. doi: 10.1016/j.cclet.2015.05.037
S.C. Weber, A.J. Spakowitz, J.A. Theriot, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 7338–7343. doi: 10.1073/pnas.1119505109
L.V. Zingman, A.E. Alekseev, D.M. Hodgson-Zingman, A. Terzic, J. Appl. Physiol. 103 (2007) 1888–1893. doi: 10.1152/japplphysiol.00747.2007
J.M. Qin, X. Li, W. Lang, et al., Chin. Chem. Lett. 35 (2024) 108925. doi: 10.1016/j.cclet.2023.108925
P.D. Boyer, Biochemistry 26 (1987) 8503–8507. doi: 10.1021/bi00400a001
H. Sigel, R. Griesser, Chem. Soc. Rev. 34 (2005) 875–900. doi: 10.1039/b505986k
X. Zhou, X. Wang, L. Shang, Chin. Chem. Lett. 34 (2023) 108093. doi: 10.1016/j.cclet.2022.108093
J.A. Cruz-Aguado, Y. Chen, Z. Zhang, et al., J. Am. Chem. Soc. 126 (2004) 6878–6879. doi: 10.1021/ja0490424
K.T. Bush, S.H. Keller, S.K. Nigam, J. Clin. Invest. 106 (2000) 621–626. doi: 10.1172/JCI10968
C. Song, Y. Xiao, K. Li, et al., Chin. Chem. Lett. 30 (2019) 1249–1252. doi: 10.1016/j.cclet.2019.03.055
A. Surprenant, R.J. Evans, Nature 362 (1993) 211–212.
H. Sigel, N.A. Corfù, Eur. J. Biochem. 240 (1996) 508–517. doi: 10.1111/j.1432-1033.1996.0508h.x
R.J. Evans, V. Derkach, A. Surprenant, Nature 357 (1992) 503–505. doi: 10.1038/357503a0
C. Li, R.W. Peoples, Z. Li, F.F. Weight, Proc. Natl. Acad. Sci. U. S. A. 90 (1993) 8264–8267. doi: 10.1073/pnas.90.17.8264
A. Sorrenti, J. Leira-Iglesias, A. Sato, T.M. Hermans, Nat. Commun. 8 (2017) 15899–15906. doi: 10.1038/ncomms15899
S. Maiti, I. Fortunati, C. Ferrante, et al., Nat. Chem. 8 (2016) 725–731. doi: 10.1038/nchem.2511
A. Mishra, D.B. Korlepara, M. Kumar, et al., Nat. Commun. 9 (2018) 1295–1303. doi: 10.1038/s41467-018-03542-z
S. Dhiman, A. Jain, M. Kumar, S.J. George, J. Am. Chem. Soc. 139 (2017) 16568–16575. doi: 10.1021/jacs.7b07469
S. Dhiman, A. Jain, S.J. George, Angew. Chem. Int. Ed. 56 (2017) 1329–1333. doi: 10.1002/anie.201610946
H. Wang, Z. Feng, B. Xu, Chem. Soc. Rev. 46 (2017) 2421–2436. doi: 10.1039/C6CS00656F
J.H.K.K. Hirschberg, L. Brunsveld, A. Ramzi, et al., Nature 407 (2000) 167–170. doi: 10.1038/35025027
G.D. Pantoş, P. Pengo, J.K.M. Sanders, Angew. Chem. Int. Ed. 46 (2007) 2138 -2138. doi: 10.1002/anie.200790048
H.J. Kim, W.C. Zin, M. Lee, J. Am. Chem. Soc. 126 (2004) 7009–7014. doi: 10.1021/ja049799v
S. Shin, S. Lim, Y. Kim, et al., J. Am. Chem. Soc. 135 (2013) 2156–2159. doi: 10.1021/ja400160j
G. Yuan, C. Zhu, Y. Liu, et al., J. Am. Chem. Soc. 131 (2009) 10452–10460. doi: 10.1021/ja901154p
L. Brunsveld, B.G.G. Lohmeijer, J.A.J.M. Vekemans, E.W. Meijer, Chem. Commun. (2000) 2305–2306.
F.J.M. Hoeben, P. Jonkheijm, E.W. Meijer, A.P.H.J. Schenning, Chem. Rev. 105 (2005) 1491–1546. doi: 10.1021/cr030070z
F. Würthner, C.R. Saha-Möller, B. Fimmel, et al., Chem. Rev. 116 (2015) 962–1052.
Y. Pu, Y. Li, X. Jin, et al., Mol. Cell 73 (2019) 143–156. doi: 10.1016/j.molcel.2018.10.022
A. Patel, L. Malinovska, S. Saha, et al., Science 356 (2017) 753–756. doi: 10.1126/science.aaf6846
L. Heinen, A. Walther, Sci. Adv. 5 (2019) eaaw0590. doi: 10.1126/sciadv.aaw0590
X. Hao, W. Sang, J. Hu, Q. Yan, ACS Macro Lett. 6 (2017) 1151–1155. doi: 10.1021/acsmacrolett.7b00649
J. Deng, A. Walther, J. Am. Chem. Soc. 142 (2020) 685–689. doi: 10.1021/jacs.9b11598
C. Pezzato, L.J. Prins, Nat. Commun. 6 (2015) 7790–7797. doi: 10.1038/ncomms8790
Y. Kang, C. Wang, K. Liu, et al., Langmuir 28 (2012) 14562–14566. doi: 10.1021/la303271f
C. Pezzato, B. Lee, K. Severin, L.J. Prins, Chem. Commun. 49 (2013) 469–471. doi: 10.1039/C2CC38058G
L. Liang, Y. Chen, X.M. Chen, et al., Chin. Chem. Lett. 29 (2018) 989–991. doi: 10.1016/j.cclet.2017.12.022
G. von Maltzahn, D.H. Min, Y. Zhang, et al., Adv. Mater. 19 (2007) 3579–3583. doi: 10.1002/adma.200701183
C. Qin, Y. Li, Q. Li, et al., Chin. Chem. Lett. 32 (2021) 3531–3534. doi: 10.1016/j.cclet.2021.05.006
C. Wang, Q. Chen, Z. Wang, X. Zhang, Angew. Chem. Int. Ed. 49 (2010) 8612–8615. doi: 10.1002/anie.201004253
J. Liu, M. Morikawa, N. Kimizuka, J. Am. Chem. Soc. 133 (2011) 17370–17374. doi: 10.1021/ja2057924
H. Duan, F. Cao, M. Zhang, et al., Chin. Chem. Lett. 33 (2022) 2459–2463. doi: 10.1016/j.cclet.2021.11.010
G. Das, P. Talukdar, S. Matile, Science 298 (2002) 1600–1602. doi: 10.1126/science.1077353
J. Deng, W. Liu, M. Sun, A. Walther, Angew. Chem. Int. Ed. 61 (2022) e202113477. doi: 10.1002/anie.202113477
E.S. Priyanka, S.K. Brar, et al., Chem. Sci. 13 (2022) 274–282. doi: 10.1039/D1SC05353A
W.C. Liao, C.H. Lu, R. Hartmann, et al., ACS Nano 9 (2015) 9078–9086. doi: 10.1021/acsnano.5b03223
X. He, Y. Zhao, D. He, et al., Langmuir 28 (2012) 12909–12915. doi: 10.1021/la302767b
C.L. Zhu, C.H. Lu, X.Y. Song, et al., J. Am. Chem. Soc. 133 (2011) 1278–1281. doi: 10.1021/ja110094g
M. Zhao, Y. Zhang, S. Yuan, et al., Soft Matter 15 (2019) 3655–3658. doi: 10.1039/C9SM00480G
R. Mo, T. Jiang, R. DiSanto, et al., Nat. Commun. 5 (2014) 3364. doi: 10.1038/ncomms4364
M. Yu, S. Jambhrunkar, P. Thorn, et al., Nanoscale 5 (2013) 178–183. doi: 10.1039/C2NR32145A
R. Mo, T. Jiang, W. Sun, Z. Gu, Biomaterials 50 (2015) 67–74. doi: 10.1016/j.biomaterials.2015.01.053
W.H. Chen, X. Yu, W.C. Liao, et al., Adv. Funct. Mater. 27 (2017) 1702102. doi: 10.1002/adfm.201702102
S. Levi, D. Finazzi, Front. Pharmacol. 5 (2014) 99.
R.H. Olesen, T.M. Hyde, J.E. Kleinman, et al., Transl. Psychiaty 6 (2016) e838. doi: 10.1038/tp.2016.83
A. Rembach, D.J. Hare, M. Lind, et al., Int. J. Alzheimers Dis. 2013 (2013) 623241.
J. Xu, S.J. Church, S. Patassini, et al., Metallomics 9 (2017) 1106–1119. doi: 10.1039/C7MT00074J
Alzheimer's association calcium hypothesis workgroup, Z.S. Khachaturian, Alzheimers Dement 13 (2017) 178–182 e17. doi: 10.1016/j.jalz.2016.12.006
Z. Yao, B. Huang, X. Hu, et al., Analyst 138 (2013) 1649–1652. doi: 10.1039/c3an00151b
S. Roy, V.S.S. Adury, A. Rao, et al., Angew. Chem. Int. Ed. 61 (2022) e202203924. doi: 10.1002/anie.202203924
W. Sun, G. Liu, M. Tong, et al., Analyst 146 (2021) 1892–1896. doi: 10.1039/D0AN02256J
S. Biswas, K. Kinbara, T. Niwa, et al., Nat. Chem. 5 (2013) 613–620. doi: 10.1038/nchem.1681
S. Dhiman, A. Sarkar, S.J. George, RSC Adv. 8 (2018) 18913-18925. doi: 10.1039/C8RA03225D
B.A. Grzybowski, K. Fitzner, J. Paczesny, S. Granick, Chem. Soc. Rev. 46 (2017) 5647–5678. doi: 10.1039/C7CS00089H
F. Pu, J. Ren, X. Qu, Chem. Soc. Rev. 47 (2018) 1285–1306. doi: 10.1039/C7CS00673J
Y. Jia, J. Li, Nat. Rev. Chem. 3 (2019) 361–374. doi: 10.1038/s41570-019-0100-8
Y. Wu, J. Wen, H. Li, et al., Chin. Chem. Lett. 28 (2017) 1916–1924. doi: 10.1016/j.cclet.2017.09.032
J. Fei, J. Li, Curr. Opin. Colloid Interface Sci. 63 (2023) 101647. doi: 10.1016/j.cocis.2022.101647
D. White, Q. Yang, Cells 11 (2022) 1920. doi: 10.3390/cells11121920
J. Deng, A. Walther, Adv. Mater. 32 (2020) 2002629. doi: 10.1002/adma.202002629
W.N. Lipscomb, N. Sträter, Chem. Rev. 96 (1996) 2375–2434. doi: 10.1021/cr950042j
A. Mishra, S. Dhiman, S.J. George, Angew. Chem. Int. Ed. 60 (2021) 2740– 2756. doi: 10.1002/anie.202006614
O. Kennard, N.W. Isaacs, W.D.S. Motherwell, et al., Proc. R. Soc. Lond. A 325 (1971) 401–436. doi: 10.1098/rspa.1971.0177
C. Li, M. Numata, M. Takeuchi, S. Shinkai, Angew. Chem. 117 (2005) 6529–6532. doi: 10.1002/ange.200501823
M. Morikawa, M. Yoshihara, T. Endo, N. Kimizuka, J. Am. Chem. Soc. 127 (2005) 1358–1359. doi: 10.1021/ja043844h
V.W.W. Yam, V.K.M. Au, S.Y.L. Leung, Chem. Rev. 115 (2015) 7589–7728. doi: 10.1021/acs.chemrev.5b00074
J.A.G. Williams, A. Beeby, E.S. Davies, et al., Inorg. Chem. 42 (2003) 8609–8611. doi: 10.1021/ic035083+
Y. Ai, Y. Li, M.H.Y. Chan, et al., J. Am. Chem. Soc. 143 (2021) 10659–10667. doi: 10.1021/jacs.1c04200
M.C.L. Yeung, V.W.W. Yam, Chem. Soc. Rev. 44 (2015) 4192–4202. doi: 10.1039/C4CS00391H
Y. Ai, M.H.Y. Chan, A.K.W. Chan, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 13856–13861. doi: 10.1073/pnas.1908034116
Z. Gao, Y. Han, Z. Gao, F. Wang, Acc. Chem. Res. 51 (2018) 2719–2729. doi: 10.1021/acs.accounts.8b00340
B. Li, Y. Li, M.H.Y. Chan, V.W.W. Yam, J. Am. Chem. Soc. 143 (2021) 21676–21684. doi: 10.1021/jacs.1c10943
M.J. Sun, Y. Liu, W. Zeng, et al., J. Am. Chem. Soc. 141 (2019) 6157–6161. doi: 10.1021/jacs.9b02055
V.W.W. Yam, A.K.W. Chan, E.Y.H. Hong, Nat. Rev. Chem. 4 (2020) 528–541. doi: 10.1038/s41570-020-0199-7
Y. Ai, Y. Fei, Z. Shu, et al., Chem. Eng. J. 450 (2022) 138390. doi: 10.1016/j.cej.2022.138390
M.C.L. Yeung, V.W.W. Yam, Chem. Sci. 4 (2013) 2928–2935. doi: 10.1039/c3sc50383f
H. Fan, K. Li, T. Tu, et al., Angew. Chem. Int. Ed. 61 (2022) e202200727. doi: 10.1002/anie.202200727
M. Kumar, O.A. Ushie, S.J. George, Chem. Eur. J. 20 (2014) 5141–5148. doi: 10.1002/chem.201304106
M. Kumar, S.J. George, Chem. Sci. 5 (2014) 3025–3030. doi: 10.1039/C4SC00813H
X. Zhao, K.S. Schanze, Chem. Commun. 46 (2010) 6075–6077. doi: 10.1039/c0cc01332c
Z. Li, R. Acharya, S. Wang, K.S. Schanze, J. Mater. Chem. C 6 (2018) 3722–3730. doi: 10.1039/C7TC05081J
Z. Li, C.J. Zeman, S.R. Valandro, et al., J. Am. Chem. Soc. 141 (2019) 12610–12618. doi: 10.1021/jacs.9b04133
J.V. Hollingsworth, A.J. Richard, M.G.H. Vicente, P.S. Russo, Biomacromolecules 13 (2011) 60–72.
K. Venkata Rao, D. Miyajima, A. Nihonyanagi, T. Aida, Nat. Chem. 9 (2017) 1133–1139. doi: 10.1038/nchem.2812
S. Ogi, K. Sugiyasu, S. Manna, et al., Nat. Chem. 6 (2014) 188–195. doi: 10.1038/nchem.1849
F. Helmich, C.C. Lee, M.M.L. Nieuwenhuizen, et al., Angew. Chem. 122 (2010) 4031–4034. doi: 10.1002/ange.201000162
T. Fukui, S. Kawai, S. Fujinuma, et al., Nat. Chem. 9 (2017) 493–499. doi: 10.1038/nchem.2684
M.F.J. Mabesoone, A.J. Markvoort, M. Banno, et al., J. Am. Chem. Soc. 140 (2018) 7810–7819. doi: 10.1021/jacs.8b02388
M. Kumar, M.D. Reddy, A. Mishra, S.J. George, Org. Biomol. Chem. 13 (2015) 9938–9942. doi: 10.1039/C5OB01448D
M. Kumar, N. Jonnalagadda, S.J. George, Chem. Commun. 48 (2012) 10948–10950. doi: 10.1039/c2cc35438a
M. Kumar, P. Brocorens, C. Tonnelé, et al., Nat. Commun. 5 (2014) 5793–5801. doi: 10.1038/ncomms6793
Y. Li, Q. Li, X. Miao, et al., Angew. Chem. Int. Ed. 60 (2021) 6744–6751. doi: 10.1002/anie.202012681
V. Stumpf, K. Gokhberg, L.S. Cederbaum, Nat. Chem. 8 (2016) 237–241. doi: 10.1038/nchem.2429
D. Zhen, C. Liu, Q. Deng, et al., Chin. Chem. Lett. 35 (2024) 109249. doi: 10.1016/j.cclet.2023.109249
C. Andreini, I. Bertini, G. Cavallaro, et al., J. Biol. Inorg. Chem. 13 (2008) 1205–1218. doi: 10.1007/s00775-008-0404-5
R. Strick, P.L. Strissel, K. Gavrilov, R. Levi-Setti, J. Cell Biol. 155 (2001) 899–910. doi: 10.1083/jcb.200105026
B. Wu, C.A. Davey, J. Mol. Biol. 398 (2010) 633–640. doi: 10.1016/j.jmb.2010.03.038
B.P. Zietz, H.H. Dieter, M. Lakomek, et al., Sci. Total Environ. 302 (2003) 127–144. doi: 10.1016/S0048-9697(02)00399-6
D. Giri, S.K. Raut, C.K. Behera, S.K. Patra, Polymer 253 (2022) 124951–124962. doi: 10.1016/j.polymer.2022.124951
L. Zhang, R. Buchet, G. Azzar, Biophys. J. 86 (2004) 3873–3881. doi: 10.1529/biophysj.103.034116
S.H. Jung, K.Y. Kim, J.H. Lee, et al., ACS Appl. Mater. Interfaces 9 (2017) 722–729. doi: 10.1021/acsami.6b12857
X. Xu, L. Feng, J. Li, et al., Chin. Chem. Lett. 30 (2019) 549–552. doi: 10.1016/j.cclet.2018.11.026
R. Mogaki, P.K. Hashim, K. Okuro, T. Aida, Chem. Soc. Rev. 46 (2017) 6480–6491. doi: 10.1039/C7CS00647K
C. Pezzato, D. Zaramella, M. Martinelli, et al., Org. Biomol. Chem. 13 (2015) 1198–1203. doi: 10.1039/C4OB02052A
C. Yu, W. Zhu, Z. He, et al., Colloids Surf. A Physicochem. Eng. Aspects 615 (2021) 126255. doi: 10.1016/j.colsurfa.2021.126255
H. Cho, Y.Y. Cho, M.S. Shim, et al., BBA Mol. Basis Dis. 1866 (2020) 165808. doi: 10.1016/j.bbadis.2020.165808
Y. Jiang, H. Zhou, W. Zhao, S. Zhang, Anal. Chem. 94 (2022) 6771–6780. doi: 10.1021/acs.analchem.2c00409
X. Cong, J. Chen, R. Xu, Front. Bioeng. Biotech. 10 (2022) 916952. doi: 10.3389/fbioe.2022.916952
X. Wen, M. Yang, L. Li, et al., Chin. Chem. Lett. 35 (2024) 109291. doi: 10.1016/j.cclet.2023.109291
L. Li, J. Wang, H. Jiang, et al., Chin. Chem. Lett. 34 (2023) 107506. doi: 10.1016/j.cclet.2022.05.020
L. Li, S. Li, J. Wang, et al., Chin. Chem. Lett. 34 (2023) 108399. doi: 10.1016/j.cclet.2023.108399
J. Pang, X. Feng, Q. Liang, et al., ACS Nano 16 (2022) 4175–4185. doi: 10.1021/acsnano.1c10029
K. Jiang, P. Jing, H. Luo, X. Cao, Mater. Chem. Phys. 289 (2022) 126460. doi: 10.1016/j.matchemphys.2022.126460
Q. Guo, Y. Liu, Z. Wang, et al., Acta Biomater. 122 (2021) 343–353. doi: 10.1016/j.actbio.2020.12.052
J. Chen, Y. Zhang, L. Zhao, et al., ACS Appl. Mater. Interfaces 13 (2021) 53564–53573. doi: 10.1021/acsami.1c14385
E. Sameiyan, E. Bagheri, S. Dehghani, et al., Acta Biomater. 123 (2021) 110–122. doi: 10.1016/j.actbio.2020.12.057
Y. Wang, X. Shang, J. Liu, Y. Guo, Talanta 176 (2018) 652–658. doi: 10.1016/j.talanta.2017.08.087
W.C. Liao, Y.S. Sohn, M. Riutin, et al., Adv. Funct. Mater. 26 (2016) 4262– 4273. doi: 10.1002/adfm.201600069
Y. Zhang, Y. Lu, F. Wang, et al., Small 13 (2017) 1602494. doi: 10.1002/smll.201602494
R. Mo, T. Jiang, Z. Gu, Angew. Chem. Int. Ed. 53 (2014) 5815–5820. doi: 10.1002/anie.201400268
H. Li, S. Zhuang, Y. Yang, et al., Int. J. Biol. Macromol. 183 (2021) 839–851. doi: 10.1016/j.ijbiomac.2021.05.020
C. Qian, Y. Chen, S. Zhu, et al., Theranostics 6 (2016) 1053–1064. doi: 10.7150/thno.14843
Z. Jiang, Y. Wang, L. Sun, et al., Biomaterials 197 (2019) 41–50. doi: 10.1016/j.biomaterials.2019.01.001
Z. Zhou, M. Vázquez-González, I. Willner, Chem. Soc. Rev. 50 (2021) 4541–4563. doi: 10.1039/D0CS01030H
W.H. Chen, W.C. Liao, Y.S. Sohn, et al., Adv. Funct. Mater. 28 (2018) 1705137. doi: 10.1002/adfm.201705137
X.R. Song, S.H. Li, H. Guo, et al., Adv. Sci. 5 (2018) 1801201. doi: 10.1002/advs.201801201
Y. Yuan, C. Du, C. Sun, et al., Nano Lett. 18 (2018) 921–928. doi: 10.1021/acs.nanolett.7b04307
J. Chen, Y. Zhang, Y. Zhang, et al., Chin. Chem. Lett. 32 (2021) 3034–3038. doi: 10.1016/j.cclet.2021.03.079
W. Feng, C. Feng, B. Wang, et al., Mater. Today Commun. 25 (2020) 101455. doi: 10.1016/j.mtcomm.2020.101455
C. Qi, Y.J. Zhu, B.Q. Lu, et al., Small 10 (2014) 2047–2056. doi: 10.1002/smll.201302984

Figure 1 Summary of ATP-induced small molecules assembly, optical properties and sensing applications.

Figure 2 (a) Chemical structural features and 3D energy minimum structure of ATP. (b) Chemical structure and binding modes of ATP receptor groups.

Figure 3 (a) Emission spectral changes of 10 upon increasing ATP concentration. Reprinted with permission [90]. Copyright 2013, Royal Society of Chemistry. (b) left: UV–vis spectra of 11; right: Photographs of 11/ATP under (Top) visible and (Bottom) UV light. Reprinted with permission [91]. Copyright 2022, Wiley-VCH Verlag GmbH. Co.KCaA.Weinheim. (c) UV–vis spectra of 12 through nucleation-growth self-assembly mechanism induced by ATP. Reprinted with permission [21]. Copyright 2018, Springer Nature. (d, e) Different methods A and B of CT formation between molecules 8 and 13 driven by ATP. Reprinted with permission [92]. Copyright 2014, John Wiley and Sons.

Figure 4 (a) The molecular structure of 14 and the probably schematic morphologies transformation of PDPA-assembly inducing by ATP. Reprinted with permission [93]. Copyright 2014, Royal Society of Chemistry. (b) The UV–vis absorption (left) and emission (right) changes during the transformations form H1 to H2 upon ATP binding. Reprinted with permission [93]. Copyright 2014, Royal Society of Chemistry. (c) The molecular structure of 15 and images of 15 and 15-ATP water solution under UV (365 nm) and sunlight illumination. Reprinted with permission [96]. Copyright 2019, American Chemical Society. (d) The UV–vis absorption (left) and emission spectra (right) of 15 in water upon increasing ATP concentrations. Reprinted with permission [96]. Copyright 2019, American Chemical Society.

Figure 5 (a, b) CD spectra changes to characterize the transformation of 14 from H1 to H2 aggregates upon ATP titration. Reprinted with permission [93]. Copyright 2014, Royal Society of Chemistry. (c) The CD curves at 495 nm upon gradually addition of different kinds of phosphates. Reprinted with permission [103]. Copyright 2015, Royal Society of Chemistry. (d) Above: the scheme of helicity reversal in architectures of 14 through binding competition of AMP, ADP and ATP; below: The chiral behaviors of molecule 8 adopted a majority rules using ATP and ADP mixtures. Reprinted with permission [105]. Copyright 2014, Springer Nature.

Figure 6 (a) The scheme of the supramolecular helical conversion of 8 controlled by the competitive electrostatic interaction between ATP and ADP. Reprinted with permission [23]. Copyright 2017, John Wiley and Sons. (b) Overlaid spectra of CD changes of the consecutively (red curve) and simultaneously (blue curve) added ATP and 12 to the pre-grown seed. Reprinted with permission [21]. Copyright 2018, Springer Nature. (c) Chemical structure of 17, and schemes of the preparation of SOF-17 via homodimerization between the coumarin moieties in 17 and cucurbit[8]uril (CB[8]) and adaptive chirality of SOF-17 induced by ATP. Reprinted with permission [106]. Copyright 2023, John Wiley and Sons. (d) Scheme of self-assembly and disassembly of 11 with glum = 0.20 induced by ATP and apyrase, respectively. Reprinted with permission [91]. Copyright 2022, John Wiley and Sons.

Figure 7 (a) Scheme of the preparation of the visual probe and the mechanism to detect Cu(Ⅱ). Reprinted with permission [63]. Copyright 2013, Royal Society of Chemistry. (b) Scheme of the selectivity and luminescent switch of 18 with ATP and Cu(Ⅱ). Reprinted with permission [65]. Copyright 2021, Royal Society of Chemistry. (c) Visualized luminescence changes of 19 for detecting Fe(Ⅲ). Reprinted with permission [113]. Copyright 2022, Elsevier. (d) The selective detection of Ca(Ⅱ) by AuNP/ATP particles. Reprinted with permission [64]. Copyright 2022, John Wiley and sons.

Figure 8 (a) Scheme of the disassembly of 10 under ATPase-catalyzed reaction of ATP. Reprinted with permission [90]. Copyright 2013, Royal Society of Chemistry. (b) Scheme of the assembly/disassembly of 15 induced by ATP and ALP, respectively. Reprinted with permission [96]. Copyright 2019, American Chemical Society. (c) Scheme of enzymatic action on the transformation of (P)-NDPA-ATP, (M)-NDPA-ADP and (rac)-NDPA-Pi. Reprinted with permission [105]. Copyright 2014, Springer Nature. (d) Scheme of nucleation-growth and seeded assembly of 12 and ATP under enzymatically modulation. Reprinted with permission [21]. Copyright 2018, Springer Nature. (e) Conformation conversion of 8 controlled by CPK and HK. Reprinted with permission [23]. Copyright 2016, John Wiley and Sons. (f) Scheme of the trypsin (Trp) enzymatic activity modulated by GumBAn and ATP. Reprinted with permission [117]. Copyright 2017, Royal Society of Chemistry.

Figure 9 (a) Left: the mechanism diagram of Dox release from ATP-responsive nanocarrier conformational transformation; Right: fluorescence spectrum of released Dox from ATP concentration-dependent Dox/Duplex. Reprinted with permission [54]. Copyright 2014, Springer Nature. (b) Left: ATP-responsive mechanism diagram of releasing Dox and Rhodamine 6G from hydrogel-coated NMOF; Right: Contrast fluorescence spectra of a blank control (1), other analogues TTP (2), GTP (3), CTP (4), and ATP (5) triggered release Dox. Reprinted with permission [139]. Copyright 2017, John Wiley and Sons. (c) Schematic diagram of ATP triggering QDs@TA-PEG/Dox dissociation assembly to release Dox. Reprinted with permission [140]. Copyright 2018, John Wiley and Sons. (d) The mechanism diagram of ATP in cancer cells triggering the binding of GroEL-Dox to the target protein plectin, leading to the release of Dox by GroEL conformational conversion. Reprinted with permission [141]. Copyright 2018, American Chemical Society.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: