Citation:
Yueying Yang, Huiru Xie, Xinbo Yu, Yang Liu, Hui Wang, Hua Li, Lixia Chen. Design, synthesis and evaluation of the first DYRK1A degrader for promoting the proliferation of pancreatic β-cells[J]. Chinese Chemical Letters,
2024, 35(11): 109570.
doi:
10.1016/j.cclet.2024.109570
Design, synthesis and evaluation of the first DYRK1A degrader for promoting the proliferation of pancreatic β-cells
English
Design, synthesis and evaluation of the first DYRK1A degrader for promoting the proliferation of pancreatic β-cells
Fujian Key Laboratory of Chinese Materia Medica, Institute of Structural Pharmacology & TCM Chemical Biology, College of Pharmacy, Fujian University of Traditional Chinese Medicine, Fuzhou 350122, China
b.
Wuya College of Innovation, Key Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China
syzyclx@163.com (L. Chen). 1 These authors contributed equally to this work.
Received Date:
07 November 2023 Accepted Date:
26 January 2024 Revised Date:
24 January 2024 Available Online:
15 November 2024
Abstract:
Dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) is the most promising target for diabetes treatment by promoting β-cell proliferation. The desmethylbellidifolin (DMB) as a DYRK1A inhibitor could facilitate β-cell proliferation in vivo and in vitro. However, DMB has the problem of weak binding affinity to DYRK1A, which means that continuous high concentration administration of DMB is effective for the diabetes. In order to solve this problem, we designed and synthesized a series of DMB-based proteolysis targeting chimeras (PROTACs) by taking advantage of the property of PROTAC that induce protein degradation in a cycle-catalytic manner. MDM2-based PROTAC X1–4P-MDM2 was identified as the most active PROTAC molecule. Mechanism research showed that X1–4P-MDM2 formed a ternary complex with DYRK1A and murine double minute 2 (MDM2), and induced the degradation of DYRK1A through the ubiquitin-proteasome system pathway. At a dose much lower than that of DMB, X1–4P-MDM2 still significantly enhanced β-cell proliferation by inhibiting transforming growth factor beta (TGF-β) and promoting the mitogen-activated protein kinases/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway, which may provide a new strategy for the application of DMB in diabetes.
Marked β-cell deficiency and dysfunction resulting in inadequate insulin production is a major pathogenesis of type 1 diabetes mellitus (T1DM) [1,2]. Traditionally, type 2 diabetes mellitus (T2DM) is considered to be caused by insulin resistance in key target organs such as the liver, adipocytes and skeletal muscle. In recent years, β-cell deficiency and dysfunction have been found to play a synergistic role with insulin resistance in the etiology of T2DM [3-5]. Therefore, reducing β-cell death or damage, inducing β-cell differentiation, and promoting β-cell proliferation have become promising strategies for the treatment of T1DM and T2DM [6,7], which creates opportunities for drug discovery in diabetes treatment by expanding and maintaining β-cell mass.
A number of biological targets have been investigated in exploring β-cell proliferation, including dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) [8,9], Erb3 binding protein-1 (EBP1) [10] and glycogen synthase kinase 3β (GSK3β) [11,12], etc. DYRK1A, as a negative regulator of the calcineurin-nuclear factor of activate T-cells (NFAT) signaling pathway, causes the inhibition of β-cell proliferation by phosphorylating the NFATc protein [8,13-15]. Targeted inhibition of DYRK1A serves as a promising strategy to induce β-cell proliferation by promoting activation of the calcineurin/NFAT signaling pathway. The development of DYRK1A inhibitors as a potential strategy for diabetes treatment has been widely reported, including harmine and analogues [15,16], 5-iodotubericidin [17], 1,3,4-thiadiazines [18], aminopyrazines (GNF-4877) [11], CC-401 [19], 1,5-naphthridines [20], leucettine-L41 [21] and desmethylbellidifolin (DMB) [22], etc. DMB was identified as a DYRK1A inhibitor through high-throughput screening in our previous study. By analyzing the co-crystal structure of the complex of DMB and DYRK1A, we have clarified the molecular mechanism of DMB in diabetes treatment [22]. However, as a small molecule compound derived from natural products, DMB has the problem of weak binding affinity to DYRK1A (Kd = 5.11 ± 0.33 µmol/L), which means that continuous high-concentration and high dosing frequency administration of DMB is effective for the diabetes. This greatly limits the application of DMB in diabetes treatment.
Proteolysis targeting chimeras (PROTACs) have garnered significant attention due to their ability to reduce cellular levels of protein of interest (POI) through E3-ligase-dependent ubiquitination [23]. In particular, PROTAC harnesses the endogenic E3 ubiquitin ligase to degrade POI in a cycle-catalytic manner [24-26], which means that ligands with weak affinity can also degrade target proteins in the form of PROTAC at lower concentrations. Therefore, applying PROTAC technology to DMB may be a feasible solution for DMB to be used in diabetes treatment.
Here, we designed 28 PROTAC degraders by covalently conjugating X1 (DMB analogue) with six different ubiquitin ligase ligands known to recruit cereblon (CRBN), murine double minute 2 (MDM2), von Hippel-Lindau (VHL), and a cellular inhibitor of apoptosis (cIAP). Fortunately, we discovered a DYRK1A degrader, X1–4P-MDM2, with a potent enzyme inhibitory activity and degradation activity. Furthermore, X1–4P-MDM2 promoted the proliferation of INS-1 cells by degrading DYRK1A. Importantly, the effect of promoting INS-1 cell proliferation after treatment with 2 µmol/L X1–4P-MDM2 was similar to that of 75 µmol/L DMB, which means that applying PROTAC to DMB can solve the high-dose problem of DMB to a certain extent. Moreover, our work developed a potent DYRK1A degrader and revealed for the first time Nutlin-3 as E3 ligase ligand to induce DYRK1A degradation. Overall, this strategy of DYRK1A degradation holds promise for further applications of DMB in treatment of diabetes.
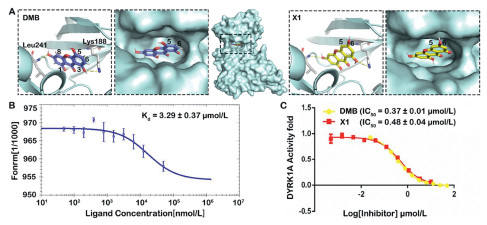
In the early stage, we isolated a series of benzochromone compounds from the plant of Swertia pseudochinensis, and evaluated their DYRK1A enzyme inhibitory activity. Among them, DMB has significant inhibitory activity on DYRK1A. By analyzing the co-crystal structure of the complex of DMB and DYRK1A (PDB ID: pdb:6LN1) [22], it was found that the 3,8-hydroxyl groups of DMB are essential active groups to form hydrogen bond with DYRK1A Lys188 and Leu241. In addition, it was found that the 5-hydroxyl group and the 6-carbon atom of the benzene ring are located at the pocket outlet (Fig. 1A). Due to the difficulty in obtaining DMB, we envisioned designing and synthesizing a new ligand X1 for DYRK1A through chemical means (Scheme S1 in Supporting information). X1 has a similar binding affinity to DMB (DMB Kd = 5.11 ± 0.33 µmol/L, X1 Kd = 3.29 ± 0.37 µmol/L) (Fig. 1B), X1 also has a similar DYRK1A enzyme inhibitory activity to DMB (DMB half effective inhibitory concentration (IC50) = 0.37 ± 0.01 µmol/L, X1 IC50 = 0.48 ± 0.04 µmol/L) (Fig. 1C), which is conducive to the large-scale synthesis of degradation agents.
Figure 1
Figure 1.
Selection of connection site and affinity test of DYRK1A ligand. (A) Schematic diagram and close-up view of the DMB or X1 in DYRK1A binding pocket. DMB was shown as purple sticks; X1 was shown as yellow sticks; Lys188 and Leu241 were shown as gray sticks. (B) Measurement of binding affinity of X1 to DYRK1A by microscale thermophoresis (MST). (C) Measurement of enzyme inhibitory activity of DMB and X1 to DYRK1A. Data are the mean of three experiments and are expressed as mean ± standard deviation (SD).
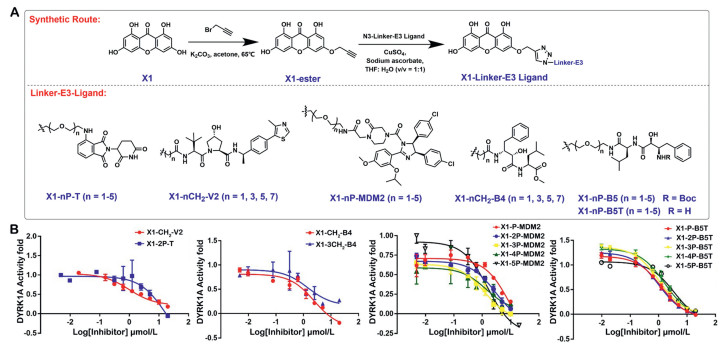
The DMB-DYRK1A co-crystal structure showed that the 3-hydroxyl group of DMB forms a hydrogen bond with Lys188, and the 8-hydroxyl group forms a hydrogen bond with Leu241, while the 5-hydroxyl group (DMB) and 6-hydroxyl group (X1) are pointing towards the solvent [22]. These observations suggested that 6-hydroxyl group possible vectors for the PROTAC linkage (Fig. 1A). Therefore, we selected the 6-hydroxyl group as the connection site for the linker-E3 ligands. Two kinds of linker polyethylene glycol (P) and carbon chains (C) were chosen as the start materials. Considering the influence of linker length and E3 ligase ligands on PROTAC binding activity and pharmacokinetic properties [27], we chose nine linker variants (1–5P, 1C, 3C, 5C, 7C) and five E3 ligase ligands to design and synthesize a series of PROTACs, corresponding to four E3 ligases, thalidomide 4-fluoride (CRBN ligand) [28,29], VH032 (VHL ligand) [30], Nutlin-3 (MDM2 ligand) [31], B4 and B5 (cIAP ligand) (Fig. 2A) [32]. The structures of X1-Linker-E3 ligands were shown in Table S1.
Figure 2
Figure 2.
Design and synthesis of DYRK1A degraders. (A) The overview of synthesizing DYRK1A-linker-E3 ligand. (B) IC50 curves of PROTACs for DYRK1A enzyme inhibitory activity. Data are the mean of three experiments and are expressed as mean ± SD.
We first evaluate the DYRK1A enzyme inhibitory activity of 28 synthesized DYRK1A-PROTACs, using DMB and X1 as positive control. A primary screening for 20 µmol/L DYRK1A-PROTACs was performed and more than half of the PROTACs inhibited DYRK1A above 75% (Table S2 in Supporting information). Subsequently, we selected PROTACs with superior inhibition of DYRK1A activity and determined their IC50 (Table S3 in Supporting information). Among them, MDM2-based and cIAP-based PROTACs exhibited DYRK1A inhibitory activity similar to DMB and X1. Notably, the enzyme inhibitory activity of MDM2-based PROTACs gradually increased with the extension of the linker length. Among all the cIAP-based PROTACs, the inhibitory activity of cIAP(B5T)-based PROTACs was better than that of the cIAP(B4)-based and cIAP(B5)-based PROTACs. Except for X1–5P-B5T, all cIAP(B5T)-based PROTACs showed strong inhibitory activity on DYRK1A (Fig. 2B).
We evaluated the DYRK1A degradation efficiency of all synthesized PROTACs at 6.7 and 20 µmol/L in INS-1 cells. The results showed that CRBN-based, VHL-based and cIAP(B4)-based PROTAC had almost no degradation potency for DYRK1A at 6.7 and 20 µmol/L (Figs. S1A, D and E in Supporting information), which was basically consistent with the evaluation results of enzyme inhibitory activity. Surprisingly, although cIAP(B5)-based PROTAC does not have stronger enzyme inhibitory activity compared to cIAP(B5T), some PROTACs exhibit obvious degradation activity at 20 µmol/L and the degradation potency gradually increases as the linker is extended. Contrary to cIAP(B5)-based PROTAC, cIAP(B5T)-based PROTACs enzyme inhibitory activity is equivalent to DMB but has almost no DYPK1A degradation potency. Among all PROTACs, MDM2-based PROTACs have the best degradation efficiency (about 40%–80%) and gradually increases with the extension of the linker. Among them, X1–4P-MDM2 and X1–5P-MDM2 have been found as remarkable DYRK1A degradation. Both of them reduced the DYRK1A protein level by more than 50% at 20 µmol/L after 24 h treatment (Figs. S1F and S2 in Supporting information). They showed potent degradation with half maximal degradation concentration in 48 h (DC50, 48 h) = 2.28 µmol/L, maximum level of target degradation in 48 h (Dmax, 48 h) = 82.84% and DC50, 48 h = 15.24 µmol/L, Dmax, 48 h = 60.24%, respectively. Notably, X1–4P-MDM2 also has significant inhibitory activity against DYRK1A. Taken together, the above results encouraged us to further characterize X1–4P-MDM2.
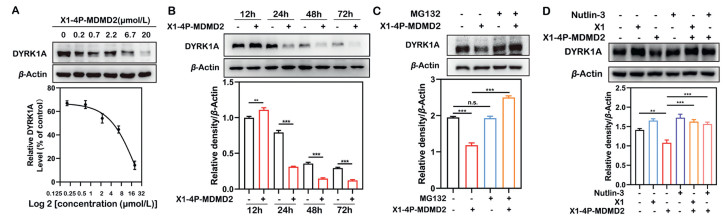
We further evaluated X1–4P-MDM2 for its DYRK1A degradation kinetics in the INS-1 cells. The results showed that X1–4P-MDM2 induced DYRK1A degradation in a concentration-dependence and time-dependence manner, which effectively reduced the DYRK1A protein within 24 h with 6.7 µmol/L treatment (Figs. 3A and B). To examine if X1–4P-MDM2 functions as a bona-fide PROTAC degrader in the INS-1 cell line, we performed several mechanistic studies using inhibitors to block protein degradation pathways. In order to examine whether X1–4P-MDM2 exerts degradation potency through the ubiquitin proteasome system (UPS), we used the proteasome inhibitor MG132 to pretreat INS-1 cells and found that it could block the degradation of DYRK1A by X1–4P-MDM2 (Fig. 3C). These results demonstrated that X1–4P-MDM2 induced the DYRK1A degradation through the proteasome. Furthermore, we treated INS-1 cells with X1 or Nutlin-3 (MDM2 ligand) alone and found that neither treatment degraded DYRK1A (Fig. 3D). However, pre-treatment with X1 or Nutlin-3, the degradation induced by X1–4P-MDM2 was abrogated (Fig. 3D), indicating that X1–4P-MDM2 is required to form a ternary complex with DYRK1A and MDM2 as an integral event in triggering DYRK1A degradation. In addition, the cellular thermal shift assay (CETSA) was employed to validate the interaction between X1–4P-MDM2 and DYRK1A. Treatment of INS-1 cells with X1–4P-MDM2 significantly improved the thermal stability of the DYRK1A protein compared with the DMSO group (Fig. S3 in Supporting information). Collectively, our findings suggested that X1–4P-MDM2 induced DYRK1A degradation by UPS pathway.
Figure 3
Figure 3.
X1–4P-MDM2 induced DYRK1A degradation via the ubiquitin-proteasome system. (A) Immunoblot analysis of DYRK1A in INS-1 cells treated with X1–4P-MDM2 at the indicated concentration for 24 h. (B) X1–4P-MDM2 degraded DYRK1A in a time-dependent manner in INS-1 cells at 6.7 µmol/L. (C) Immunoblot analysis of DYRK1A protein in INS-1 cells pre-treated with MG132 (1 µmol/L) for 4 h and subsequently treated with either DMSO or X1–4P-MDM2 (20 µmol/L) for 24 h. (D) Pre-treatment with X1 (40 µmol/L) or Nutlin-3 (60 µmol/L) for 4 h blocked DYRK1A degradation by X1–4P-MDM2 (20 µmol/L). Statistical significance was calculated with unpaired two-tailed Student's t-test. **P < 0.01, ***P < 0.001 vs. the X1–4P-MDM2 treated group. n.s., not significant.
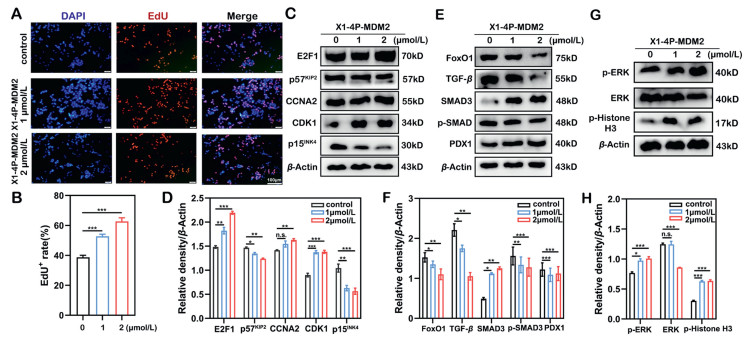
Considering that X1–4P-MDM2 has a strong ability on degrading DYRK1A, we used Cell-Light™ EdU assay to further explore the effect of X1–4P-MDM2 on INS-1 cell proliferation. The results showed that the percentage of EdU+ positive in INS-1 cells increased after X1–4P-MDM2 treatment, indicating that low concentrations X1–4P-MDM2 can promote the proliferation of INS-1 cells (Figs. 4A and B).
Figure 4
Figure 4.
Evaluation of the ability of X1–4P-MDM2 to promote INS-1 proliferation. (A, B) EdU assay was performed to measure the proliferation capacity of INS-1 cells after 24 h administration of X1–4P-MDM2. (C, D) The effect of X1–4P-MDM2 on the expression level of INS-1 cell cycle proteins (E2F1, p57KIP2, CDK1, CCNA2 and p15INK4). (E, F) The effect of X1–4P-MDM2 on the expression of FoxO1, TGF-β, SMAD3, p-SMAD3 and PDX1, was measured after 24 h. (G, H) The expression of ERK, p-ERK and p-histone H3 after X1–4P-MDM2 treatment was analyzed by Western blot. Data are the mean of three experiments and are expressed as mean ± SD. Statistical significance was calculated with unpaired two-tailed Student's t-test. P < 0.05, **P < 0.01, ***P < 0.001 vs. the X1–4P-MDM2 treated group.
To explore the mechanism by which X1–4P-MDM2 promotes cell proliferation, INS-1 cells were treated with 1 and 2 µmol/L X1–4P-MDM2 for 24 h, and cell cycle downstream factors and proliferation-related signaling pathways were evaluated. The results showed that X1–4P-MDM2 induced the decrease of cell cycle inhibitory factors p15INK4 and p57KIP2, and upregulated the expression of CDK1 and E2F1 to promote INS-1 cell proliferation (Figs. 4C and D). According to previous studies, transforming growth factor beta/small mothers against decapentaplegic (TGF-β/SMAD) and mitogen-activated protein kinases/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway are mainly involved in promoting DMB-mediated islet β-cell proliferation [22]. Therefore, we evaluated the effect of X1–4P-MDM2 on these two signaling pathways. X1–4P-MDM2 treatment resulted in a reduction of phosphorylated SMAD3 and an upregulation of SMAD3. In addition, the expression of PDX1 increased, by contrast, the expression of forkhead box protein O1 (FoxO-1) and TGF-β decreased after X1–4P-MDM2 treatment (Figs. 4E and F). An increase in phosphorylated ERK and phosphorylated histone H3 also found in X1–4P-MDM2 treatment of INS-1 cells (Figs. 4G and H). Collectively, our findings indicate that X1–4P-MDM2 inhibits TGF-β and promotes the MAPK/ERK signaling pathway to promote β-cell proliferation, which is consistent with the mechanism of DMB.
DYRK1A is a negative regulator of the calcineurin/NFAT signaling pathway. Inhibiting DYRK1A protein expression can promote the activation of the calcineurin/NFAT signaling pathway, thereby promoting β-cell proliferation [14,33]. There is growing evidence that blocking DYRK1A activity or reducing its expression has become a feasible approach for diabetes treatment [34]. The DYRK1A inhibitor DMB promotes β-cell proliferation in vivo and in vitro. However, due to the weak affinity between DMB and DYRK1A, problems with high dosage and high frequency of administration occur in the actual medication process, which might limit the application of DMB in diabetes treatment. Traditional small molecules and antibodies inhibit the function of target proteins through an "occupancy-driven" model to treat diseases. This mode requires inhibitors or monoclonal antibodies with high affinity or concentration to occupy the active site of the target and block the transduction of downstream signaling pathways. However, PROTAC exerts its activity through "event-driven", which does not affect the function of the POI, but mediates the degradation of the disease-causing POI [35,36]. Therefore, PROTAC harnesses the ubiquitin-proteasome system to degrade POI in a cycle-catalytic manner, suggesting that it can work below the catalyst amount. Applying PROTAC technology to DMB may solve the problems of high dosage and high frequency of DMB administration to a certain extent.
Here, we synthesized and evaluated a series of DYRK1A degraders utilizing X1 (DMB analogue) and six E3 ligase ligands. Among these compounds, X1–4P-MDM2 displayed the most potent activity in decreasing DYRK1A levels (DC50, 48 h = 2.28 µmol/L, Dmax, 48 h = 82.84%). Furthermore, we elucidated X1–4P-MDM2 induced DYRK1A degradation by forming a ternary complex with MDM2 and DYRK1A and ultimately activating the ubiquitin-proteasome system pathway. Importantly, X1–4P-MDM2 (2 µmol/L) promoted the expression of cell cycle-related proteins and activated the MAPK/ERK and TGF-β signaling pathways by degrading DYRK1A at doses much lower than DMB (75 µmol/L), thereby promoting INS-1 cell proliferation. By converting a DYRK1A inhibitor into a DYRK1A PROTAC, we developed a low-dose and high-efficiency DYRK1A-targeting pro-proliferation agent of pancreatic β-cells. These findings may have important clinical implications for the treatment of diabetes.
In summary, we developed a DYRK1A degrader using PROTAC to solve the problem of high-dose treatment of DMB. This not only eliminates the limitations of DMB in diabetes treatment but also broadens the application scope of PROTAC to a certain extent. Meanwhile, our study presented insights into the design and study of degraders targeting DYRK1A.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors thank the National Natural Science Foundation of China (NSFC, No. 82141216), Chunhui Program-Cooperative Research Project of the Ministry of Education, Liaoning Province Natural Science Foundation (No. 2022-MS-241), Shenyang Young and Middle-aged Innovative Talents Support Program (No. RC210446), “Select the best candidates to lead key research projects” of Fujian University of Traditional Chinese Medicine (No. XJB2022008), and Foundation of Fujian University of Traditional Chinese Medicine (No. X2023001-Talent) for financial supports. And we acknowledged the support from National-Local Joint Engineering Research Center for Molecular Biotechnology of Fujian & Taiwan TCM, at Fujian University of Traditional Chinese Medicine.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109570.
[1]
M. von Herrath, M. Peakman, B. Roep, Clin. Exp. Immunol. 172 (2013) 186–202. doi: 10.1111/cei.12085
[2]
M.L. Campbell-Thompson, M.A. Atkinson, A.E. Butler, et al., Diabetologia 56 (2013) 2541–2543. doi: 10.1007/s00125-013-3043-5
[3]
P.W. Franks, E. Pearson, J.C. Florez, Diabetes Care 36 (2013) 1413–1421. doi: 10.2337/dc12-2211
[4]
A.E. Butler, J. Janson, S. Bonner-Weir, et al., Diabetes 52 (2003) 102–110. doi: 10.2337/diabetes.52.1.102
[5]
J. Rahier, Y. Guiot, R.M. Goebbels, C. Sempoux, J.C. Henquin, Diabetes Obes. Metab. 10 (Suppl 4) (2008) 32–42. doi: 10.1111/j.1463-1326.2008.00969.x
[6]
A. Vetere, A. Choudhary, S.M. Burns, B.K. Wagner, Nat. Rev. Drug. Discov. 13 (2014) 278–289. doi: 10.1038/nrd4231
[7]
P. Wang, N.M. Fiaschi-Taesch, R.C. Vasavada, et al., Nat. Rev. Endocrinol. 11 (2015) 201–212. doi: 10.1038/nrendo.2015.9
[8]
E. Dirice, D. Walpita, A. Vetere, et al., Diabetes 65 (2016) 1660–1671. doi: 10.2337/db15-1127
[9]
Y. Ogawa, Y. Nonaka, T. Goto, et al., Nat. Commun. 1 (2010) 86. doi: 10.1038/ncomms1090
[10]
W. Shen, M.S. Tremblay, V.A. Deshmukh, et al., J. Am. Chem. Soc. 135 (2013) 1669–1672. doi: 10.1021/ja309304m
Figure 1
Selection of connection site and affinity test of DYRK1A ligand. (A) Schematic diagram and close-up view of the DMB or X1 in DYRK1A binding pocket. DMB was shown as purple sticks; X1 was shown as yellow sticks; Lys188 and Leu241 were shown as gray sticks. (B) Measurement of binding affinity of X1 to DYRK1A by microscale thermophoresis (MST). (C) Measurement of enzyme inhibitory activity of DMB and X1 to DYRK1A. Data are the mean of three experiments and are expressed as mean ± standard deviation (SD).
Figure 2
Design and synthesis of DYRK1A degraders. (A) The overview of synthesizing DYRK1A-linker-E3 ligand. (B) IC50 curves of PROTACs for DYRK1A enzyme inhibitory activity. Data are the mean of three experiments and are expressed as mean ± SD.
Figure 3
X1–4P-MDM2 induced DYRK1A degradation via the ubiquitin-proteasome system. (A) Immunoblot analysis of DYRK1A in INS-1 cells treated with X1–4P-MDM2 at the indicated concentration for 24 h. (B) X1–4P-MDM2 degraded DYRK1A in a time-dependent manner in INS-1 cells at 6.7 µmol/L. (C) Immunoblot analysis of DYRK1A protein in INS-1 cells pre-treated with MG132 (1 µmol/L) for 4 h and subsequently treated with either DMSO or X1–4P-MDM2 (20 µmol/L) for 24 h. (D) Pre-treatment with X1 (40 µmol/L) or Nutlin-3 (60 µmol/L) for 4 h blocked DYRK1A degradation by X1–4P-MDM2 (20 µmol/L). Statistical significance was calculated with unpaired two-tailed Student's t-test. **P < 0.01, ***P < 0.001 vs. the X1–4P-MDM2 treated group. n.s., not significant.
Figure 4
Evaluation of the ability of X1–4P-MDM2 to promote INS-1 proliferation. (A, B) EdU assay was performed to measure the proliferation capacity of INS-1 cells after 24 h administration of X1–4P-MDM2. (C, D) The effect of X1–4P-MDM2 on the expression level of INS-1 cell cycle proteins (E2F1, p57KIP2, CDK1, CCNA2 and p15INK4). (E, F) The effect of X1–4P-MDM2 on the expression of FoxO1, TGF-β, SMAD3, p-SMAD3 and PDX1, was measured after 24 h. (G, H) The expression of ERK, p-ERK and p-histone H3 after X1–4P-MDM2 treatment was analyzed by Western blot. Data are the mean of three experiments and are expressed as mean ± SD. Statistical significance was calculated with unpaired two-tailed Student's t-test. P < 0.05, **P < 0.01, ***P < 0.001 vs. the X1–4P-MDM2 treated group.

 DownLoad:
DownLoad:

 下载:
下载:





