Key Laboratory of Synthetic and Natural Functional Molecule of the Ministry of Education, Xi'an Key Laboratory of Functional Supramolecular Structure and Materials, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, China
yfhan@nwu.edu.cn (Y.-F. Han). 1 These authors contributed equally to this work.
Received Date:
04 November 2023 Accepted Date:
26 December 2023 Revised Date:
03 December 2023 Available Online:
15 November 2024
Abstract:
Conventionally, organic radicals adhere to the Aufbau principle, the energy level of the singly occupied molecular orbital (SOMO) is not below the highest occupied molecular orbital (HOMO), but somewhat abnormal phenomena have appeared recently. In this study, we introduce a novel strategy by incorporating unique NHC-Au-X units into a tris(2,4,6-trichlorophenyl)methyl (TTM) system to create metal-involved open-shell complexes, denoted as TTM-NHC-Au-X (X = I, Br, or Cl). Density-functional theory calculations were used to predict an inversion in the energy of the SOMO and highest doubly occupied molecular orbital (HOMO) of TTM-NHC-Au-I, which is supported by experimental results. Organometallic radicals TTM-NHC-Au-X demonstrated distinct properties with different coordinated halides. The radical behaviors have been investigated by EPR, UV–vis spectroscopy and cyclic voltammetry, additional structural information provided by structurally comparing related the precursor complexes given by X-ray crystallography. TTM-NHC-Au-I with SOMOHOMO conversion (SHC) features a highly thermal decomposition temperature up to 305 ℃. Furthermore, the photostability of TTM-NHC-Au-I was found to be 75 and 23 times greater than that of TTM-NHC-Au-Br and TTM-NHC-Au-Cl, respectively. These findings provide valuable insights into the structural and electronic design principles governing the occurrence of SOMOHOMO conversion in open-shell systems.
An energetic inversion of the singly occupied molecular orbital (SOMO) and the highest doubly occupied molecular orbital (HOMO) level is termed SOMO—HOMO conversion (SHC) [1–7]. The radicals with peculiar orbital energetics contribute their unique electronic properties, where the SOMO is not the highest occupied orbital in the species [8–11]. Recent extensive research on non-Aufbau electronic structures has led to conceptual developments in the switch for bond dissociation energy, transforming certain radical species into high-spin states through one-electron oxidation [5,8–16]. Specifically, SHC is associated with the unusual photostability or thermostability of organic radicals [12,13,15], and several SHC radicals exhibit high luminescence quantum yields, offering possibilities for their application in organic light-emitting diode devices [15,16]. SHC systems have been mainly achieved in organic radical molecules [17–28]. SHCs triggered by coordination to the metal center have been mostly limited in TEMPO-bound (TEMPO = 2,2,6,6-tetramethylpiperidine-l-oxy radical) dithiolate ligands [4,29,30]. Related metal-involved species are far less common, presumably because the methods that allow efficient control of SHC within metal complexes remain rare.
In this study, we describe a method for generating SHC within a series of NHC-Au (NHC = N-heterocyclic carbene) complexes based on molecular design. We focused on combining tris(2,4,6-trichlorophenyl)methyl (TTM) moiety and Au-NHC-halide units to realize a unique electronic structure by selecting coordination halides at the metal center. A series of radical-based functional materials have been achieved using the covalent attachment of different skeletons to TTM radicals [15,16,31–45]. Note that SHC could be performed when the HOMO orbital of the peripheral substituent is higher than the SOMO orbital of the TTM, and vice versa [46].
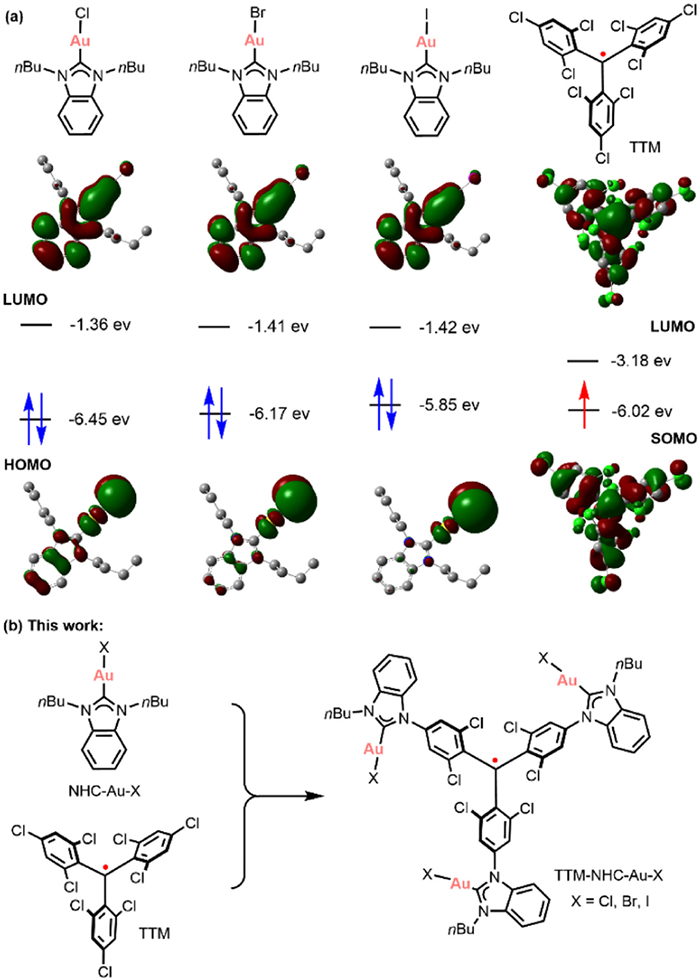
NHC-Au-X complexes exhibit interesting properties attributed to the characteristic electronic structure with different donating abilities of the halide ligands (X = I, Br or Cl) [47,48]. Although they have been rather frequently applied as catalysts in many organic reactions [49–54], their use in radical material systems is uncommon. Based on the density-functional theory (DFT) calculation results, the HOMO and LUMO energy levels of NHC-Au-X complexes depend significantly on the electron-donating ability of the halide ligands (Fig. 1). The energy of the SOMO of TTM (−6.02 eV) is lower than the HOMO of NHC-Au-I (−5.85 eV), but higher than those in NHC-Au-Br (−6.17 eV) and NHC-Au-Cl (−6.45 eV). We believe that by introducing a unique NHC-Au-X unit in the TTM system, new classes of materials with SHC properties can be readily obtained through facile modification of coordinated halide atoms.
Figure 1
Figure 1.
(a) Spatial distribution of the frontier molecular orbitals and corresponding energy levels of NHC-Au-X (X = I, Br or Cl) and TTM calculated via DFT methods [(M06, UM06)/SDD (Au, Br, I) and 6–31G* (H, C, N, Cl)]. (b) The schematic synthesis of TTM-NHC-Au-X (X = I, Br or Cl).
This study, successfully synthesized a series of TTM-centered gold carbene radicals TTM-NHC-Au-X (X = I, Br or Cl), and their precursors αHTTM-NHC-Au-X. The electronic structure, photophysical properties, electrochemical studies, and stability of these newly obtained organometallic radicals were investigated, along with their single-crystal X-ray diffraction (XRD) analysis. The iodine atoms on the gold centers allow for an SHC phenomenon, that tunes the property of the molecule. However, SHC was not observed when the bromide or chloride was used as coordinated ligands. Notably, TTM-NHC-Au-I showed significantly higher thermal and photostability than its Br/Cl counterparts.
According to our molecular design, three NHC precursors, benzimidazolium-substituted triarymethyl compounds 2a-c were designed and synthesized from tris[4-(1H-benzimidazolyl)−2,6-dichlorophenyl]methyl (1) following Scheme 1. Treatment of 2a-c with [AuCl(THT)] to generate trinuclear gold(I) complexes αHTTM-NHC-Au-X (3a-c) in anhydrous dichloromethane under the exclusion of light in good yields. Compounds 2a-c and 3a-c were verified using nuclear magnetic resonance (NMR) spectroscopy (1H and 13C{1H}) and high-resolution electrospray ionization (HR-ESI) mass spectrometry (Figs. S1-S27 in Supporting information). In the 13C NMR spectrum of complexes 3a-c, which displays decreasing upfield shifts of the CNHC resonance from 187.4 ppm (3a) > 181.4 ppm (3b) > 178.1 ppm (3c). These different chemical shifts reveal the characteristic donating ability of halide atoms, where the more upfield shift corresponds to weaker donor fragments [47,48]. Subsequently, complexes 3a-c were treated with tetrabutylammonium hydroxide (TBAH) in anhydrous THF to generate the corresponding anions. Furthermore, through oxidation with tetrachloro-p-chloranil, three organometallic radical complexes 4a-c containing different halogen atoms were isolated after workup (Scheme 1). The complexes 4a-c were characterized through HR-ESI, element analysis, ultraviolet-visible (UV–vis) adsorption spectroscopy, and single-crystal XRD analysis (4b and 4c). The radical feature in 4a-c was confirmed using electron paramagnetic resonance (EPR) spectroscopy.
Scheme 1
Scheme 1.
Synthesis of ligands 2a-c, complexes 3a-c and radicals 4a-c. (i) nBuBr or nBuI, DMF, 120 ℃, 36 h; (ii) NH4PF6, MeOH, r.t., 24 h; (iii) K2CO3, AuCl(THT), dry DCM, in dark, r.t., 36 h; (iv) TBAH, tetrachloro-p-chloranil, dry THF, in dark, r.t., 7 h. Ligand 2b is also shown as a crystal structure. Hydrogen atoms, and molecules of the solvent of crystallization are omitted for clarity. Color code: Cl, green; Br, brown green; C, grey; N, blue.
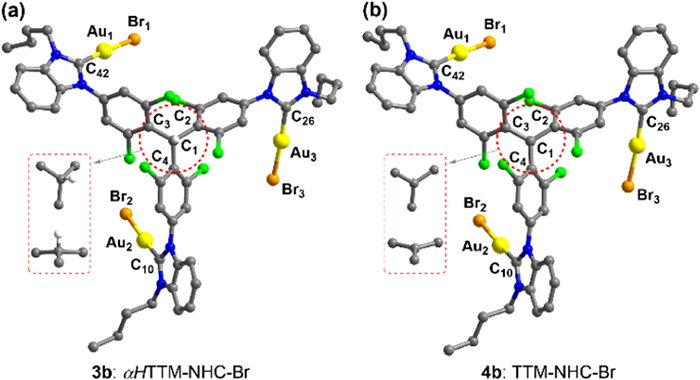
The solid-state 3a-c molecular structures were unambiguously determined through single-crystal XRD experiments (Fig. 2a and Figs. S28-S30 in Supporting information). The Au-X (X = I, Br or Cl) 3a-c bond lengths were found to decrease as the electronegativity of the halogen atoms increased, with an order of 2.532(4)−2.557(2) Å (3a) > 2.382(10)−2.396(11) Å (3b) > 2.266(2)−2.300(3) Å (3c) (Table S1 in Supporting information).
Figure 2
Figure 2.
X-ray crystal structures of (a) complex 3b and (b) radical 4b. Hydrogen atoms, the partial center structure, and molecules of the solvent of crystallization are omitted for clarity. Color code: Au, yellow; Cl, green; Br, brown green; C, grey; N, blue; H, white. Selected bond parameters in Å and deg: 3b, C1—C2 1.586(11), C1—C3 1.545(11), C1—C4 1.526(11), Au1—C42 1.999(8), Au2—C10, 1.990(8), Au3—C26, 1.976(9), Au1-Br1 2.3875(9), Au2-Br2 2.3817(10), Au3-Br3 2.3959(11), C2—C1—C3 116.5(7), C2—C1—C4 115.6(7), C3—C1—C4 118.7(8); 4b, C1—C2 1.510(10), C1—C3 1.503(10), C1—C4 1.499(10), Au1—C42 1.990(11), Au2—C10, 1.977(11), Au3—C26, 2.001(10), Au1-Br1 2.3591(13), Au2-Br2 2.3644(14), Au3-Br3 2.3939(15), C2—C1—C3 120.7(7), C2—C1—C4 119.0(7), C3—C1—C4 120.3(7).
X-ray crystallographic analyses of 4b and 4c revealed that each isolated complex consists of a TTM radical center and three NHC-Au-X (X = Br or Cl) units at the periphery (Fig. 2b, Figs. S31 and S32 in Supporting information), which agrees with the configuration inferred from HR-ESI mass spectrum in the liquid phase. Noted that the C1C2, C1C3 and C1C4 bond lengths of 4b (1.510(10) Å, 1.503(10) Å and 1.499(10) Å) are significantly shorter than those of 3b (1.586(11) Å, 1.545(11) Å and 1.526(11) Å). In contrast, the C2C1C3, C2C1C4 and C3C1C4 bond angles of 4b (120.7(7)o, 119.0(7)o and 120.3(7)o) are bigger than those of 3b (116.5(7)o, 115.6(7)o and118.7(8)o) (Fig. 2b, and Table S1 in Supporting information). Additionally, the central carbon atom C1 of radical 4b is sp2 hybridized and coplanar with three adjacent carbon atoms bonded to it (C2, C3, and C4). However, the central carbon atom C1 of complex 3b is sp3 hybridized and non-coplanar with the adjacent carbon atoms. Similar changes were observed between complexes 3c and 4c (Figs. S30 and S32, Table S1 in Supporting information). These observations revealed the radical character of organometallic complex 4b.
The neighbouring intermolecular Au…Au distances of 3b and 4b are in the range of 3.26–3.80 Å and 3.25–3.72 Å, respectively, indicating the presence of Au…Au interactions in the solid states (Figs. S29 and S31). Similar intermolecular Au…Au interactions were observed in the structures of complexes 3a, 3c and 4c (Figs. S28, S30 and S32). Additionally, intermolecular X…π interactions evidently existed in such systems (Figs. S28-S32).
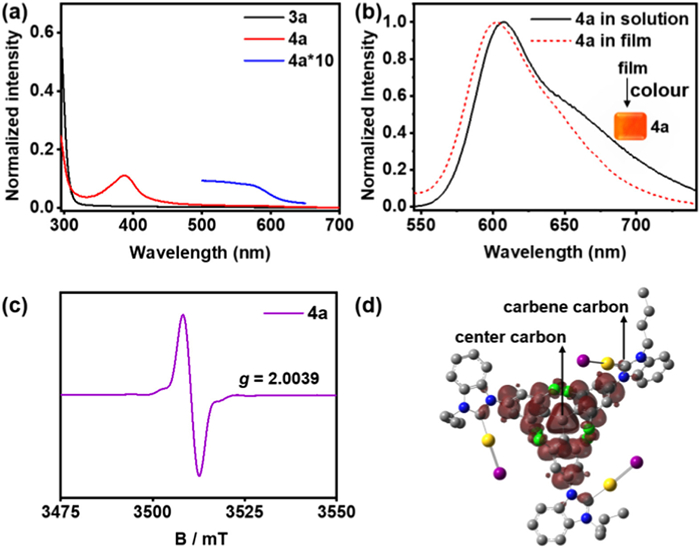
All solution samples of radical complexes 4a-c for UV–vis absorption, photoluminescence (PL) excitation and emission properties were investigated. Fig. 3a and Fig. S33 (Supporting information) show that two new characteristic absorption maxima at around 380 and 560 nm were observed after the generation of radicals in contrast to their precursor's 3a-c. There is a slight wavelength shift due to the distinct electronegativity of metal coordination atoms. For instance, a set of strong short wavelength absorption at 388 nm (4a), 380 nm (4b), 386 nm (4c), and a low-lying band at 567 nm (4a), 564 nm (4b), 563 nm (4c), respectively. For the PL spectra, the maximum fluorescence peaks were located at 592 nm (4b) and 589 nm (4c), whereas radical 4a underwent an obvious red-shift at 607 nm (average 20 nm) in toluene solution (Fig. 3b, Figs. S33d and S34 in Supporting information).
Figure 3
Figure 3.
(a) UV–vis absorption spectra of 3a and 4a in CH2Cl2 (c = 10−5 mol/L, 298 K). (b) Normalized emission spectra of 4a in PhCH3 (c = 10−4 mol/L, 298 K, solid lines) and 4a (2 wt%) doped in PMMA film (298 K, dash lines). (c) EPR spectra of 4a in CH2Cl2 (c = 10−3 mol/L, 298 K). (d) Spin density distribution of 4a calculated via DFT methods [UM06/SDD (Au, I) and 6–31G* (H, C, N, Cl)]. Color code for (d): Au, yellow; Cl, green; I, purple; C, grey; N, blue.
Complexes 4a-c were doped into poly(methyl methacrylate) (PMMA) films to investigate the photophysical properties of the radicals in solid-state thin films, and the emission spectra and quantum yields of the formed thin films were measured. In contrast to those in solution, complexes 4a (603 nm) and 4b (585 nm) exhibited slight blue shifts, and complex 4c (609 nm) has a wider red shift of 18 nm (Fig. 3b and Fig. S34 in Supporting information). The photoluminescence quantum yields (PLQY) of the spin-coated 4a-c films were 1.0% (4a), 1.8% (4b), and 1.4% (4c), respectively.
The magnetic properties of radicals 4a-c were measured at room temperature by EPR. There intense and broad EPR signal peaks with g = 2.0039 (4a), 2.0041 (4b), and 2.0044 (4c) were recorded, each featuring a typical sign of one unpaired electron (Fig. 3c and Fig. S35 in Supporting information).
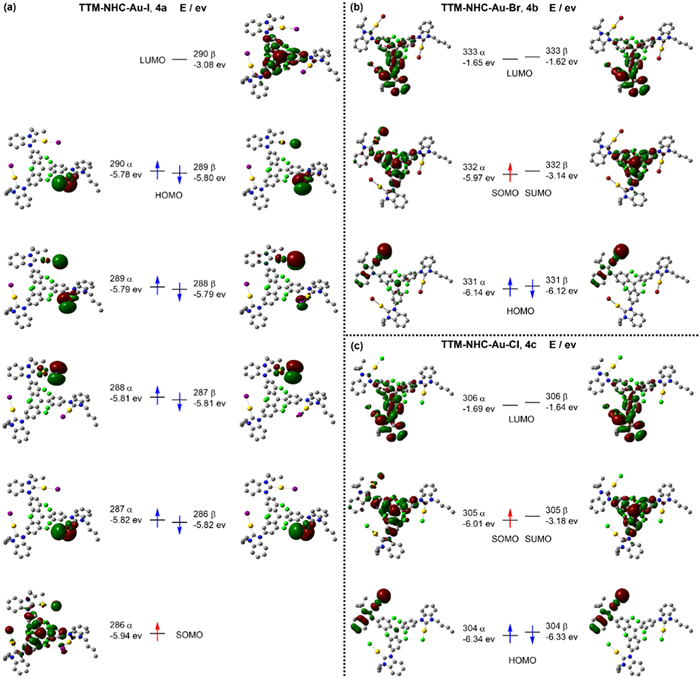
For a deeper understanding of the radical's electronic distribution and molecular orbitals, quantum chemical calculations were performed using DFT. The results demonstrate that the spin density is distributed from the center to the periphery, with the highest at the center carbon site (ca. 0.837) and a small spin density at the carbene carbon atom (ca. 0.003) (Fig. 3d and Fig. S35 in Supporting information). The molecular orbital calculation results exhibit that the SOMO of all radical complexes 4a-c is delocalized over the TTM radical unit with a minor extension to the halogen gold carbene pendants. Meanwhile, the HOMO is mainly concentrated in the halogen gold carbene segments (Fig. 4). Specifically, the SOMO orbital energy level in radical 4a is lower than that of the HOMO, indicating a clear violation of the Aufbau principle (Fig. 4a). The calculated spin density of radical 4a also reflects the SOMO orbital (286 α-spin) and the corresponding LUMO orbital (290 β-spin). Conversely, the SOMO orbitals of 4b and 4c are higher than the HOMO orbitals, which align with the Aufbau principle (Figs. 4b and c). The SOMO orbital of 4a is located in the lower energy orbital of the inner layer, which may result in lower reactivity and higher stability than 4b and 4c. We repeated the process using another method to validate the accuracy of the theoretical calculations, and the results were consistent with the expected outcomes. The DFT calculation [UB3LYP/SDD (Au, I), 6–31G* (H, C, N, Cl)] confirmed that SOMO—HOMO conversion can be achieved for 4a (Fig. S36 in Supporting information).
Figure 4
Figure 4.
Spatial distribution of the frontier molecular orbitals and corresponding energy levels of (a) 4a, (b) 4b, and (c) 4c calculated via DFT [UM06/SDD (Au, Br, I) and 6–31G* (H, C, N, Cl)].
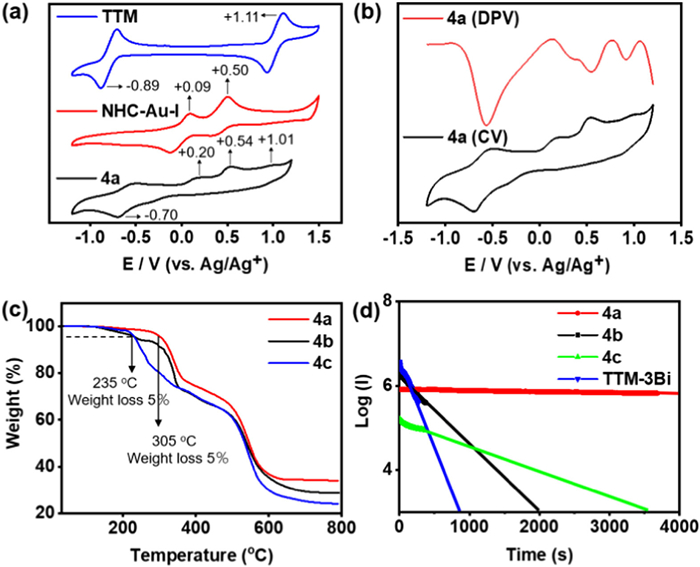
Since the redox experiment is an essential characterization method for confirming the orbital turnover of radicals [15,46], a series of redox properties were measured to gain deeper insights into the electronic characteristics of the complexes. The electrochemical behavior of the metal carbene radicals 4a-c, the radical center (TTM), and the isolated fragment molecule (NHC-Au-I) are presented graphically (Fig. 5a and Fig. S37 in Supporting information). Strikingly, the radicals 4a-c has high electrochemical stability, and no significant decomposition was observed after 50 scanning cycles. The cyclic voltammetry (CV) spectra of 4b and 4c display two pairs of redox potentials, with a reduction peak at −0.63 V (4b), −0.62 V (4c), and an oxidation peak at +1.15 V (4b), +1.16 V (4c), respectively, which correspond to the −0.89 V and +1.11 V values observed in TTM (Fig. S37). For 4a, a reduction and oxidation peak close to those of 4b and 4c appears at −0.70 V and +1.01 V. Additionally, two new oxidation peaks occur at +0.20 V and +0.54 V, which are distinct features unobserved in 4b and 4c. Notably, these new 4a peaks are excellently interconnected with the NHC-Au-I pendants, which show oxidation potentials at +0.09 V and +0.50 V (Fig. 5a). This evidence implies that 4a exhibits different redox behavior compared to 4b and 4c, further explaining their distinct electronic structures. Moreover, the differential pulse voltammogram (DPV) experiment also reveals four and two oxidation waves for radicals 4a and 4b/4c, respectively (Fig. 5b and Fig. S37). Therefore, both CV and DPV data confirm that radical 4a does not conform to the Aufbau principle, which is consistent with the DFT results.
Figure 5
Figure 5.
(a) Cyclic voltammograms of TTM (blue), NHC-Au-I (red), and 4a (black) in CH2Cl2 (nBu4NPF6, 100 mV/s, c = 10−3 mol/L). (b) Differential pulse voltammogram and Cyclic voltammogram of 4a in CH2Cl2 (nBu4NPF6, 100 mV/s, c = 10−3 mol/L). (c) Thermal gravimetric analysis (TGA) of 4a-c under N2. (d) Plots showing emission decay of 4a-c in PhCH3 under continuous excitation at λex = 380 nm (c = 10−4 mol/L, 298 K).
The thermal and photochemical stability of 4a-c were surveyed under a nitrogen atmosphere. The thermal properties of 4a-c were researched with thermogravimetric analysis (TGA). Radical 4a exhibited no degradation until attaining a temperature of 305 ℃, whereas radicals 4b and 4c had lower decomposition temperatures at approximately 235 ℃ (Fig. 5c and Fig. S38 in Supporting information). Additionally, the degeneration of their fluorescence intensity was investigated through continuous excitation at λex = 380 nm in a diluted toluene solution and compared with tris[4-(1H-benzimidazolyl)−2,6-dichlorophenyl]methyl radical (TTM-3Bi). Fig. 5d shows that the fitted half-time (t1/2) of radical 4a is 2.65 × 104 s, a value nearly two orders of magnitude larger than that of TTM-3Bi, a stronger photostability radical compound [32]. In contrast, the degenerate lifetime of the fluorescence intensity for 4b and 4c significantly decreases to 3.55 × 102 s and 1.13 × 103 s, respectively. The results above indicate that the non-Aufbau behavior exhibited by radical 4a confers superior stability compared to the Aufbau behavior observed in radicals 4b and 4c, which is consistent with previous reports for such derivatives [15].
In summary, we designed, synthesized, and characterized a new series of metal-carbene complexes of persistent TTM core-based radicals. Theoretical calculation and experiment results proved that the SOMO and HOMO orbital energy levels of radicals 4a-c could be adjusted by altering coordination halogen to metal atoms, and the organometallic radical complexes with non-Aufbau (4a) and Aufbau (4b, 4c) electronic structures were readily accessible. This strategy provides a new technique for synthesizing organometallic radical complexes that violate the Aufbau principle. Additionally, TTM-NHC-Au-I based on realizing the SOMO—HOMO converted unique electronic structure showed significantly higher thermal and photostability than its Br/Cl counterparts. Significantly improving the thermal- and photostability of radicals will expand their practical applications. Given the efficient synthetic method established in this study, different kinds of organometallic radicals with other coordinated ligands and diverse NHC skeletons can be envisioned. Thus, further research into their potential applications in multifunctional materials with optical and magnetic responses is required.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors gratefully acknowledge financial support from the National Natural Science Fund for Distinguished Young Scholars of China (No. 22025107), Shaanxi Fundamental Science Research Project for Chemistry & Biology (No. 22JHZ003), the National Youth Top-notch Talent Support Program of China, Xi'an Key Laboratory of Functional Supramolecular Structure and Materials, and the FM&EM International Joint Laboratory of Northwest University.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109465.
D. Nugegoda, N.V. Tzouras, S.P. Nolan, et al., Inorg. Chem. 61 (2022) 18802–18809. doi: 10.1021/acs.inorgchem.2c03487
[54]
P. Gao, J. Xu, T. Zhou, et al., Angew. Chem. Int. Ed. 62 (2023) e202218427. doi: 10.1002/anie.202218427
Figure 1
(a) Spatial distribution of the frontier molecular orbitals and corresponding energy levels of NHC-Au-X (X = I, Br or Cl) and TTM calculated via DFT methods [(M06, UM06)/SDD (Au, Br, I) and 6–31G* (H, C, N, Cl)]. (b) The schematic synthesis of TTM-NHC-Au-X (X = I, Br or Cl).
Scheme 1
Synthesis of ligands 2a-c, complexes 3a-c and radicals 4a-c. (i) nBuBr or nBuI, DMF, 120 ℃, 36 h; (ii) NH4PF6, MeOH, r.t., 24 h; (iii) K2CO3, AuCl(THT), dry DCM, in dark, r.t., 36 h; (iv) TBAH, tetrachloro-p-chloranil, dry THF, in dark, r.t., 7 h. Ligand 2b is also shown as a crystal structure. Hydrogen atoms, and molecules of the solvent of crystallization are omitted for clarity. Color code: Cl, green; Br, brown green; C, grey; N, blue.
Figure 3
(a) UV–vis absorption spectra of 3a and 4a in CH2Cl2 (c = 10−5 mol/L, 298 K). (b) Normalized emission spectra of 4a in PhCH3 (c = 10−4 mol/L, 298 K, solid lines) and 4a (2 wt%) doped in PMMA film (298 K, dash lines). (c) EPR spectra of 4a in CH2Cl2 (c = 10−3 mol/L, 298 K). (d) Spin density distribution of 4a calculated via DFT methods [UM06/SDD (Au, I) and 6–31G* (H, C, N, Cl)]. Color code for (d): Au, yellow; Cl, green; I, purple; C, grey; N, blue.
Figure 4
Spatial distribution of the frontier molecular orbitals and corresponding energy levels of (a) 4a, (b) 4b, and (c) 4c calculated via DFT [UM06/SDD (Au, Br, I) and 6–31G* (H, C, N, Cl)].

 DownLoad:
DownLoad:


 下载:
下载:







