Citation:
Zhenjie Yang, Chenyang Hu, Xuan Pang, Xuesi Chen. Sequence design in terpolymerization of ε-caprolactone, CO2 and cyclohexane oxide: Random ester-carbonate distributions lead to large-span tunability[J]. Chinese Chemical Letters,
2024, 35(5): 109340.
doi:
10.1016/j.cclet.2023.109340
Sequence design in terpolymerization of ε-caprolactone, CO2 and cyclohexane oxide: Random ester-carbonate distributions lead to large-span tunability
English
Sequence design in terpolymerization of ε-caprolactone, CO2 and cyclohexane oxide: Random ester-carbonate distributions lead to large-span tunability
Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
* Corresponding author. E-mail address: xpang@ciac.ac.cn (X. Pang). 1 These authors contributed equally to this work.
Received Date:
22 September 2023 Accepted Date:
23 November 2023 Revised Date:
21 November 2023 Available Online:
15 May 2024
Abstract:
It is of great interest to make a degradable material widely tailorable to replace petroleum-derived products among diverse applications. Here, we report the construction of a new multi-purpose degradable material for the first time via a simple ternary copolymerization system comprising ε-caprolactone (ε-CL), cyclohexane oxide (CHO) and CO2. Under low pressure of 1 bar ~5 bar, the ring-opening polymerization (ROP) of ε-CL and ring-opening copolymerization (ROCOP) of CO2 and CHO can simultaneously proceed. The carbonate units are randomly distributed on the polymer chain. These random terpolymers have controllable molar mass (10–106 kDa) and compositions (4–33 mol% CO2). And the obtained materials show large-span tunability from tough plastic to elastomer and even adhesive.
Aliphatic polyesters are typical degradable polymers with biocompatibility, which are used in packaging, pesticide, medicine and other fields [1-3]. However, the intrinsic nature of aliphatic main chain and lack of functionality limit the potential for high-value-added products with wider use [4]. Chemically combining different polymers seems to be a simple and effective strategy for this barrier since it enables precise tailoring of copolymer structures including sequences and compositions allowing for a much broader adjustment of properties. For a multi-component system, monomer and catalyst selection is the key to determining the reaction process and product quality [5-12]. The considerations include the ease of performing the reaction, the source and cost of the monomers, the molar masses and sequences diversification, as well as the sustainability of the resulting copolymers.
With the aid of precise synthetic approaches including ring-opening copolymerization (ROCOP) and ring-opening polymerization (ROP), the terpolymerization of ε-caprolactone (ε-CL), CO2 and epoxides appears to be a promising choice for constructing degradable materials with wide tunability [13-17]. Compared with most aliphatic polyesters, PCL is a typical soft material that has low transition temperature (−60 ~ −65 ℃) and high elongation at break (ε = 200%−1200%) [18-20]. Meanwhile, the copolymers of CO2 and CHO are brittle and could be selected as hard segments for complementarity. More importantly, the combination of PCL and poly(cyclohexane carbonate) (PCHC) leads to fully degradable materials diminishing the questions about end-of-life fate [2,3,13,18,21-24]. And copolymerization of CO2 and epoxides is an attractive way to fix CO2 [25-27]. Significant advances in the ε-CL/CO2/epoxides terpolymerization have been achieved using some metal catalysis including Zn, Mn and Cr complexes [28-33]. But the sequence diversification of the terpolymers remains challenging [34-36]. Most of them only form block- or tapered copolymers via a switchable pathway because the reactivity of CO2 is kinetically prior to that of lactones. Not to mention that most reports focused on studying switch mechanisms and improving reactivity with little exploration of the properties of products. In addition, the different reactivities of the two polymerization cycles result in two- or multi-steps reactions involving gas shift procedure. The utilization of high CO2 pressure (> 15 bar) for the CO2/epoxides ROCOP also potentially handicaps large-scale applications.
The pressure of CO2 has been proven to play a crucial role in regulating the reactivity and selectivity of lactones/CO2/CHO terpolymerization [37-40]. Rieger and coauthors reported a ternary system comprising mixed feedstock of CHO, CO2, and β-butyrolactone (BBL) with Lewis acidic zinc complex as catalyst at two different CO2 pressures. Block structures were formed at 40 bar CO2, whereas 3 bar CO2 led to the formation of statistic copolymers [39,40]. This result could be rationalized by the reduction of the CO2/epoxide coupling reaction rate to a similar rate of BBL ROP under lower CO2 pressure. Recently, we reported the synthesis of ABC triblock copolymers from anhydrides, CHO, CO2 and ε-CL mixtures via kinetic and thermodynamic regulation using a Mn catalyst [32]. Interestingly, the incorporation of CO2 into the polymer chain was achieved by the in-situ generated macro-precursors and this process was found available even under ambient CO2 pressure. Accordingly, we assume that the kinetic priority of CO2 and CHO coupling to the lactones ROP will be depressed as the CO2 pressure is reduced. The ROP of ε-CL will not be completely inhibited under low CO2 pressure and at the same time, the generated PCL chain can promote the ROCOP of CO2 and CHO (Scheme S1 in Supporting information). In this work, we develop a simple one-pot and one-step ternary copolymerization system to prepare new polyester-polycarbonate copolymers with random topologies, high molar mass and a wide range of material performance. For a one-pot and one-step terpolymerization of CO2/CHO/ε-CL, the versatile salen-Mn catalyst that can simultaneously catalyze the ROP of ε-CL and ROCOP of CO2/CHO is selected. The catalytic performance of salen-Mn catalyst in ε-CL ROP under lower CO2 pressure was evaluated. As shown in Fig. S1 (Supporting information), initial attempts, using the reported MnIIICl//bis(triphenylphosphine)iminium chloride ([PPN]Cl) catalytic system, on the ε-CL ROP under 5 bar of CO2 pressure at 80 ℃ afforded no PCL formation with only a little yield of cyclohexene carbonate. The ε-CL ROP started after the removal of CO2 and ceased when CO2 was recharged forming PCL-PCHC di-block copolymers. Further, ambient CO2 pressure was applied in the ROP under identical conditions. As determined by the 1H NMR of the crude mixture, the consumption of ε-CL was observed along with the CO2 insertion, yielding copolymers with 19.3% PCHC (Fig. S2 in Supporting information). These results indicated the feasibility of producing PCL-PCHC terpolymers under ambient CO2 pressure via a one-pot and one-step route and the potentiality for tailoring the microstructure of terpolymers by adjusting the CO2 pressure. Additionally, for comparison, a CO2/CHO coupling reaction using MnIIICl//PPNCl was conducted under ambient pressure. No polycarbonate but very little cis-cyclohexene carbonate (cis-CHC) was observed (Fig. S3 in Supporting information). This observation verified our deductions that the growing PCL chain could promote the copolymerization of CO2 and CHO even under ambient pressure.
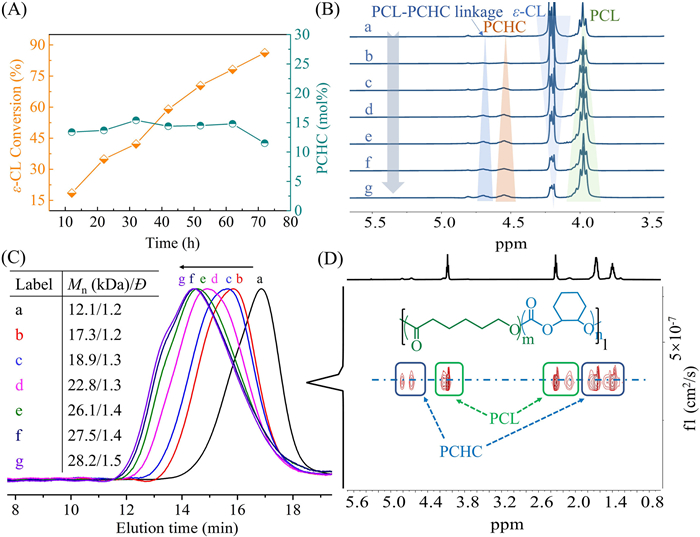
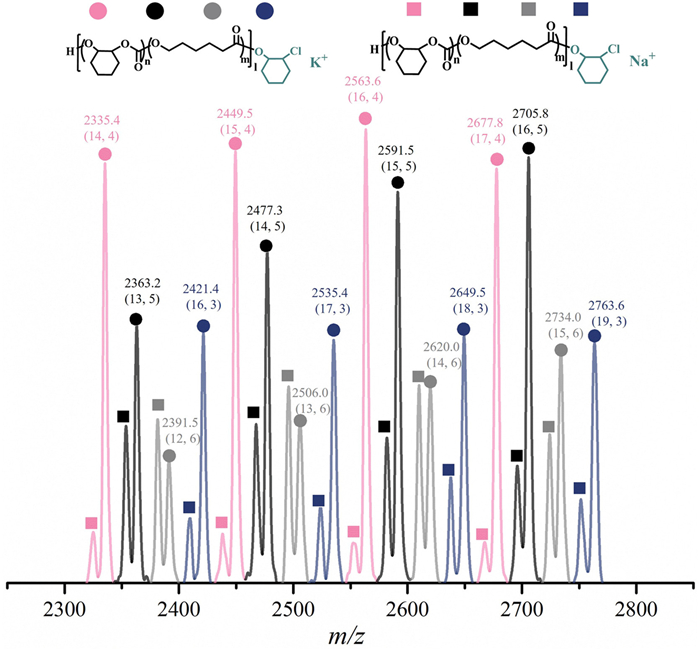
To validate the furnishing of random structures of the obtained terpolymers, the terpolymerization of ε-CL, CHO and CO2 catalyzed by an MnIIICl: [PPN]Cl ratio of 1:0.5 was monitored by NMR and size exclusion chromatography (SEC) methods at 80 ℃. Note that the optimization results of the catalyst system and the detailed assignment for characteristic signals of purified terpolymers are listed and discussed in Table S1 and Figs. S7–S10 (Supporting information). Aliquots were taken respectively at certain intervals from a one-pot reaction for analysis of conversion and molar mass. As shown in Fig. 1A, the content of PCHC was almost kept invariant (about 15%) along with the conversion of ε-CL and only dropped in the final stage which could be attributed to the hampered coupling of CO2 and CHO by the gradual viscosity of the mixture. The 1H NMR spectra showed more directly the synchronized increase in intensities at δ = 4.00 and δ = 4.55 ppm, respectively assigned to the PCL and PCHC. Notably, the intensity of resonances at δ = 4.03 and 4.70 ppm assigned to the linkage units of PCL and PCHC also increased gradually along with the reaction, which revealed the PCHC units were randomly distributed on the polymer chain rather than block structure (Fig. 1B). Besides, the SEC traces of the terpolymers exhibited increasing molar mass and relatively low Ð (Fig. 1C). And only one coefficient was observed in the DOSY spectrum of the copolymer which further evidenced that the obtained products were copolymers rather than blends (Fig. 1D and Fig. S11 in Supporting information). The analysis of MALDI-TOF-MS indicated the copolymers were all linear and α‑hydroxyl-ω-chloride end-capped (Fig. 2 and Fig. S12 in Supporting information). The detailed alignment of m/z values showed no poly(CHO) formation and the elegant combination of ROP and ROCOP.
Figure 1
Figure 1.
(A) Plots of the conversion of ε-CL and the PCHC content. (B) Time-elapsed 1H NMR spectra of ε-CL/CHO/CO2 terpolymerization, "a" to "g" denote to each aliquot collected from crude mixture after every 12 h. (C) SEC trace of the sample polymers at different ε-CL conversions corresponding to "a" to "g" in Fig. 1B. (D) The diffusion-ordered spectroscopy (DOSY) spectrum of the final polymers. cat: [PPN]Cl: ε-CL: CHO = 1:0.5:800:2000, 1 bar CO2, the reaction was conducted in 4 mL CHO under 80 ℃ and the crude mixtures were sampled every 10 h for analysis.
Figure 2.
The matrix-assisted laser desorption/ionization time-of-flight mass spectrum (MALDI-TOF-MS) analysis of low Mn copolymers collected at the initial stage of an independent one-pot copolymerization (cat: [PPN]Cl: ε-CL: CHO = 1:0.5:200:1000, 1 bar CO2, reacted for 12 h under 80 ℃). The m/z values were aligned via the equations: m × 114[ε-CL] + n × 142[CHO—CO2] + 98[CHO] + 35.5[Cl−] + 39[K+] or m × 114[ε-CL] + n × 142[CHO—CO2] + 98[CHO] + 35.5[Cl−] + 23[Na+]. The numbers in the brackets referred to the ε-CL and carbonate unit number, respectively.
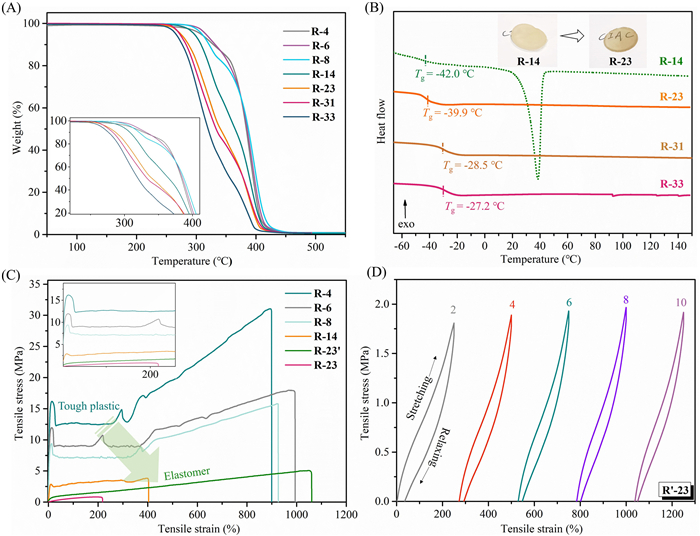
After the establishment of the optimal catalytic systems and the basic mechanism of the terpolymerization, a series of PCL-PCHC random copolymers with PCHC content from 4 mol% to 33 mol% was prepared. As listed in Table S2 (Supporting information), all reactions showed activity and the conversions of ε-CL were > 95% even at the 4000:1 feed ratio of ε-CL: catalyst. The PCHC content of the terpolymers was adjusted by shifting cocatalysts, CO2 pressure and temperature. The characterization data of these terpolymers was exhibited in Table 1. All the obtained terpolymers had high molar masses (Mn > 60 kg/mol except for R-33, and the increasing distributions might be attributed to the more transesterification reactions under higher temperatures) and exhibited excellent thermal stability (Td, 5% > 260 ℃). With the increase (from 4% to 33%) of PCHC content, the Td, 5% of the terpolymers decreased from 320 ℃ to 266 ℃. Two-stage decomposition profiles of these terpolymers were observed in Fig. 3A and the first degradation corresponded to about 10% PCL and PCHC decomposition. The second degradation occurred at about 350 ℃ corresponding to the decomposition of the remaining PCL. The Tm (from 55.6 ℃ to 38.4 ℃) and Tc (from 27.4 ℃ to 15.6 ℃) decreased significantly with the increase of PCHC content, which were getting closer to the thermal properties of PCL with low Mn (Table 2 and Fig. S26 in Supporting information). This phenomenon further indicated the random distribution of PCHC units along the polymer chains dividing the long PCL chain into short ones. Note that no obvious transition temperature was observed in both heating runs of these semi-crystalline terpolymers, partly due to the small and scattered amorphous region of the polymer. However, with the increase of PCHC segments (from R-14 to R-30), the melting regions at 38.4 ℃ disappeared accompanied by the emergence of an obvious glass transition (Fig. 3B), indicating a transformation from semi-crystalline to amorphous polymer. Thus, the solvent-cast film of R-23 turned out to be transparent while the R-14 film was optically opaque.
Table 1
Table 1.
The characterization of PCL-PCHC random terpolymers.
Figure 3.
The thermal and mechanical properties of the PCL-random-PCHC terpolymers. (A) TGA profiles of the terpolymer. Heating rate 10 ℃/min. (B) DSC thermograms of R-14 ~ R-33. Second heating runs 10 ℃/min, the traces were shifted vertically for clarity. (C) The representative stress−strain curves for uniaxial extension measurements of R-4 ~ R-23. (D) Cyclic tensile measurements for R'−23 (0–250% fixed strain, 10 cycles, 20 mm/min). Only curves for cycles 2,4,6,8 and 10 were shown. The maximum travel of the clamp was 32.75 mm and maintained for 5 s, and the minimum travel was 0 and maintained for 10 s.
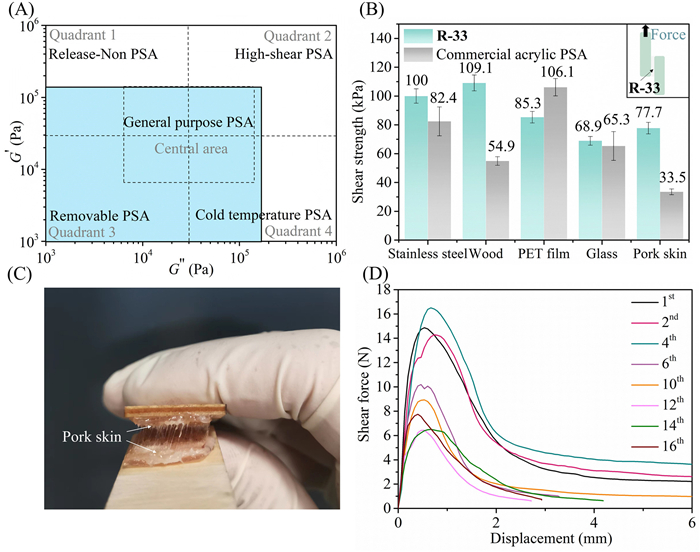
The copolymers showed broad-range tunable mechanical properties according to the tensile stress-strain curves (Fig. 3C). Firstly, with low PCHC content, the samples R-4 ~ R-14 exhibited typical behavior of ductile plastics with yield points ranging from 18% to 30% strain. Sample R-4 had the highest tensile strength (29.2 ± 2.0 MPa) and elastic modulus (306 ± 20 MPa) with high elongation at break (900% ± 40%). The slight increase of PCHC content resulted in a decline in mechanical properties. The R-6 and R-8 showed similar elongation but reduced yield point and tensile strength (17.32 ± 0.38 MPa and 13.4 ± 2.1 MPa, respectively). And the R-14 exhibited only a tensile strength of 3.9 ± 1.0 MPa with about 400% elongation at break. Notably, further increasing PCHC amount markedly changed the intrinsic nature of the copolymer. The R-23 showed no yield point with low tensile strength (< 1 MPa, and the samples quickly recover their original shapes after the tensile test (Fig. S31 in Supporting information). The R'-23 with identical composition to R-23 but higher molar mass (106.8 kDa) showed obvious elastomeric behavior like R-23 but much higher tensile strength (5.1 ± 0.5 MPa) and stretchability (1050% ± 50%) (Table S2, entry 8). It is reasonable to assume that the enhanced performance is attributed to the formation of crystalline regions by longer PCL chains in the terpolymer with higher molar mass (Fig. S32 in Supporting information). The elastomer R'-23 was further subjected to 10 reciprocating stress-strain cycles with 250% strain to evaluate the elastomeric performance. In the first cycle of tensile test, the stress-strain curves exhibited a distinct hysteresis loop with hysteresis ratio of 68.6%, as the tensile loading energy was mostly consumed by internal friction (Fig. S33 in Supporting information). After undergoing the first stretching-relaxing cycle, the subsequent hysteresis loop of R'-23 shrank significantly (Fig. 3D). The curves of cycles 4–10 were almost identical with high elastic recovery (93.5% ± 0.5%) and low residual strain (6.0% ± 0.6%). The elastic performance of sample R-23 was also tested by 10 reciprocating stress-strain cycles with 0.54 MPa stress (Fig. S34 in Supporting information). As expected, R-23 exhibited 17.0% ± 0.5% residual strain in the first cycle and 36.3% ± 0.6% after 10 cycles. The rise of extensional strain was observed along with the stretching-relaxing cycles. Interestingly, upon further increasing carbonate content to 31%, the random copolymer turned out to be a weak and soft material with a tensile strength of 0.1 ± 0.02 MPa and no obvious fracture point which could be potentially utilized as a typical adhesive (Fig. S35 in Supporting information). Oscillatory rheology analysis was performed to investigate the viscoelastic properties of R-31 (Fig. S36 in Supporting information). The shear storage modulus G′ that reflected the elasticity dominated over the loss modulus G′′(viscosity) between 1 and 102 rad/s [41,42]. A fixed oscillation at 10 rad/s vs. time experiment showed G′ (2.0 × 105 Pa) was stable and higher than the loss modulus (G′′), which indicated a soft, viscoelastic solid state of R-31 (Fig. S36A). Also, the G′ ranged from 790 Pa to 3.4 × 105 Pa lying in the Dahlquist criterion verifying the R-31 had typical rheological characteristics of pressure-sensitive adhesive (PSA). Further, the hypothetical viscoelastic window of R-31 was constructed by plotting the coordinates of (G′′, G′) at 10−1 rad/s and 102 rad/s (Fig. S36B) [41]. Most of the data points fell in Quadrant 1, revealing that R-31 might be used as high-shear PSAs. To further explore the application of these degradable poly(carbonate-b-ester) materials in PSAs for broad use, the viscoelastic properties of sample R-33, with similar PCHC content to R-31 but lower molar mass, were also probed. Different from R-31, R-33 was highly viscoelastic at room temperature (Figs. S38 and S39 in Supporting information) and was proved to be in a liquid state since the G′′ dominated over the G′ between adhesive bonding (10−2 to 1 rad/s) and debonding frequencies (102 rad/s). This result indicated that sample R-33 had higher contact efficiency than R-31. The hypothetical viscoelastic window of R-33 showed that the (G′, G′′) data points covered the full Central area and Quadrant 3, suggesting that R-33 had great potential for general-purpose PSA's even without any additives (Fig. 4A). Subsequently, the adhesive performance of R-33 to different substrates was tested using a universal testing machine. As shown in Fig. 4B, the lap shear strengths of R-33 for stainless steel, wood, PET film, glass and pork skin were 100.0, 109.1, 85.3, 68.9 and 77.7 kPa, which were comparable to, or higher than that of commercial acrylic PSA. Note that a "wiredrawing" phenomenon was observed when tearing apart two substrates like the pork skins in Fig. 4C, and the substrates could still be stuck to each other after being reattached together indicating good repeatability. Then, repeated adhering-tearing cyclic lap shear tests were performed to evaluate the reusable performance of R-33 (Fig. 4D). The pork skins were reattached again after each shear test and pressed with a 500 g weight for 10 min before the next test. The R-33 exhibited the maximum shear force of about 15 N in the first 5 cycles and sustained above 6 N after 16 adhering-tearing cycles. The promising characteristics of R-33 suggested it also could be a great candidate for repeatable medical PSAs.
Figure 4
Figure 4.
(A) Viscoelastic window for R-33 constructed from the bonding (ω = 0.1 rad/s) and debonding (ω = 100 rad/s) frequencies and only the area where G′, G′′ ≥ 103 Pa (cyan shaded area) was shown. Quadrant titles indicate PSA type. (B) The comparison of lap shear strengths of R-33 and a commercial acrylic PSA for different substrates. (C) The photograph of the "wiredrawing" phenomenon. (D) The repeatability test of R-33 as removable PSAs.
In summary, a novel degradable PCL-PCHC copolymer with random topology was synthesized with a ternary copolymerization comprising ε-CL, CHO and CO2. Via adjusting the reaction conditions, excellent control of carbonate content (4 mol% to 33 mol%) in the material was achieved. Besides high molar masses ranging from 10 kDa to 106 kDa and thermal stabilities (Td, 5% > 260 ℃), these new random terpolymers exhibited widely adjustable mechanical properties. The copolymer with 4 mol% PCHC exhibited similar tensile strength (29.2 ± 2.0 MPa) but a higher Young's modulus (306 ± 20 MPa) than pure commercial PCL (~200 MPa modulus). The increase of PCHC led to a decrease in tensile strength and the copolymer with 14 mol% PCHC possessed 3.9 ± 1.0 MPa tensile strength and 400% ± 20% strain. The copolymer with 23 mol% PCHC showed typical elastomeric behavior but lower strength (< 1 MPa). And the mechanical properties of elastomers could be significantly enhanced by increasing the molar mass. Further increasing the PCHC content to 33 mol% made the copolymer a PSA for general purpose without additives, which exhibited better adhesion ability for different substrates than a commercial acrylic PSA, as well as excellent repeatability. Overall, we believe PCL-PCHC random copolymer will prove an important strategy in the development of multifunctional degradable polymeric materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was funded by the National Key R&D Program of China (No. 2021YFA1501700), the Science and Technology Development Plan of Jilin Province (Nos. 20230101042JC, 20210201059GX), the National Natural Science Foundation of China, Basic Science Center Program (No. 51988102), the National Natural Science Foundation of China (Nos. 52203017, 52073272 and 22293062) and Bureau of International Cooperation Chinese Academy of Sciences (No. 029GJHZ2023017MI).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109340.
[1]
Y. Zhu, C. Romain, C.K. Williams, Nature 540 (2016) 354–362. doi: 10.1038/nature21001
Figure 1
(A) Plots of the conversion of ε-CL and the PCHC content. (B) Time-elapsed 1H NMR spectra of ε-CL/CHO/CO2 terpolymerization, "a" to "g" denote to each aliquot collected from crude mixture after every 12 h. (C) SEC trace of the sample polymers at different ε-CL conversions corresponding to "a" to "g" in Fig. 1B. (D) The diffusion-ordered spectroscopy (DOSY) spectrum of the final polymers. cat: [PPN]Cl: ε-CL: CHO = 1:0.5:800:2000, 1 bar CO2, the reaction was conducted in 4 mL CHO under 80 ℃ and the crude mixtures were sampled every 10 h for analysis.
Figure 2
The matrix-assisted laser desorption/ionization time-of-flight mass spectrum (MALDI-TOF-MS) analysis of low Mn copolymers collected at the initial stage of an independent one-pot copolymerization (cat: [PPN]Cl: ε-CL: CHO = 1:0.5:200:1000, 1 bar CO2, reacted for 12 h under 80 ℃). The m/z values were aligned via the equations: m × 114[ε-CL] + n × 142[CHO—CO2] + 98[CHO] + 35.5[Cl−] + 39[K+] or m × 114[ε-CL] + n × 142[CHO—CO2] + 98[CHO] + 35.5[Cl−] + 23[Na+]. The numbers in the brackets referred to the ε-CL and carbonate unit number, respectively.
Figure 3
The thermal and mechanical properties of the PCL-random-PCHC terpolymers. (A) TGA profiles of the terpolymer. Heating rate 10 ℃/min. (B) DSC thermograms of R-14 ~ R-33. Second heating runs 10 ℃/min, the traces were shifted vertically for clarity. (C) The representative stress−strain curves for uniaxial extension measurements of R-4 ~ R-23. (D) Cyclic tensile measurements for R'−23 (0–250% fixed strain, 10 cycles, 20 mm/min). Only curves for cycles 2,4,6,8 and 10 were shown. The maximum travel of the clamp was 32.75 mm and maintained for 5 s, and the minimum travel was 0 and maintained for 10 s.
Figure 4
(A) Viscoelastic window for R-33 constructed from the bonding (ω = 0.1 rad/s) and debonding (ω = 100 rad/s) frequencies and only the area where G′, G′′ ≥ 103 Pa (cyan shaded area) was shown. Quadrant titles indicate PSA type. (B) The comparison of lap shear strengths of R-33 and a commercial acrylic PSA for different substrates. (C) The photograph of the "wiredrawing" phenomenon. (D) The repeatability test of R-33 as removable PSAs.

 DownLoad:
DownLoad:


 下载:
下载:





