Figure 1.
Selected natural products with the seven-membered ring motif.
Strategies for constructing seven-membered rings: Applications in natural product synthesis
Peng Chen , Lijuan Liang , Yufei Zhu , Zhimin Xing , Zhenhua Jia , Teck-Peng Loh

Medium-sized rings, specifically seven-membered ring systems, are widely present in many important natural products and bioactive pharmaceutical compounds (Fig. 1) [1–4]. Compared to smaller [5] or larger rings, medium-sized rings have significant advantages in affinity to biological receptors, oral bioavailability, and cell permeability due to their relatively rigid and diverse 3D spatial properties [6–8]. Therefore, the synthesis and application of medium-sized ring compounds and their pharmaceutical value have been attracted great interest in both academia and industry.
Due to its special ring structure, traditional methods for constructing seven-membered carbon rings often face unfavorable entropy and enthalpy strains, resulting in limited in synthetic methods. Over the past decade, there have been significant advances in the synthesis of complex natural products with seven-membered rings. However, only few reviews were summarized by Enders [9] and Griffith [10]. In 2021, Li's group has overviewed the total synthesis of natural products involving the benzocycloheptene motifs [11]. In this review, we focus on strategies for the syntheses of natural products with cycloheptane skeletons reported from 2017 to April 2023. Various efficient synthetic strategies for synthesis of such challenging compounds have been reported, including Pauson-Khand reaction, rearrangement reactions, radical cyclization reactions, Barbier reaction, Friedel–Crafts reaction, ring-closing metathesis (RCM) reactions and intramolecular [5 + 2] cycloaddition. In this section, we will provide a brief background of each molecule and describe the synthesis of the natural product.
In 1973, Khand and Pauson reported that various ethyne hexacarbonyl dicobalt complexes reacted with olefins in hydrocarbon or ether solvents to form cyclopentenones in good yields [12,13]. Pauson and co-workers studied the applicability and limitations of this reaction in the 1970s. Typically, Pauson-Khand reaction involves a formal [2 + 2 + 1] cycloaddition of alkynes, olefins, and CO catalyzed by transition metals such as cobalt and rhodium complexes, to obtain the substituted cyclopentenone structures, which is a multicomponent reaction with high atom economy. It can be used not only for intermolecular reactions to construct five-membered rings, but also for intramolecular reactions to achieve the construction of highly strained medium-sized or large rings. Therefore, it has potential in organic synthesis, especially in the total synthesis of natural products [14–17].
Principinols C is a grayanane class diterpenoids natural product with high oxidation states, which was first isolated from the aerial part of Rhododendron principis by Hou and co-workers in 2014 [18]. Structurally, the molecule possesses an intriguing and complex 5/7/6/5 tetracylic skeleton bearing eight contiguous stereocenters. Because of its unique, highly complex structures and bioactivity, principinols C has attracted considerable attention from synthetic chemists over the past years.
Jia's group achieved the first total synthesis of (−)-principinol C, featuring an intramolecular Pauson-Khand reaction as key step to construct the 7,5-bicyclic ring system (Scheme 1) [19]. The known bicyclo [3.2.1]-octane 1 was converted to cyanide 2 in four steps involving DIBAL−H reduction, epoxidation, methoxymethyl protection and Van Leeson reaction. Ozonation of olefin 2 followed by epimerization of the C9 in one pot gave the ketone 3 in 91% yield (d.r. > 14:1). Moving forward, the Pauson-Khand reaction precursor enyne 4 was synthesized from the ketone 3 via a four steps transformation. After testing several conditions of the key Pauson-Khand reaction, treatment of enyne 4 with Co2(CO)8 at 40 ℃ gave quantitative dicobalt alkyne complex, which was converted to the desired tetracycle 5a with the increase of temperature to 60 ℃. Under the optimized condition, the key Pauson-Khand reaction provided the tetracycle 5a in 45% yield and its C6 diastereomer 5b in 9% yield, as well as the bicyclo[5.2.1]-ketone 5c in 19% yield. Then, methylation of 5a gave the intermediate 6, which was further converted to (−)-principinol C through a five steps transformation. Finally, the first total synthesis of (−)-principinol C was achieved in 16 steps with 2.8% overall yield from known bicyclo[3.2.1]octane ring system 1.

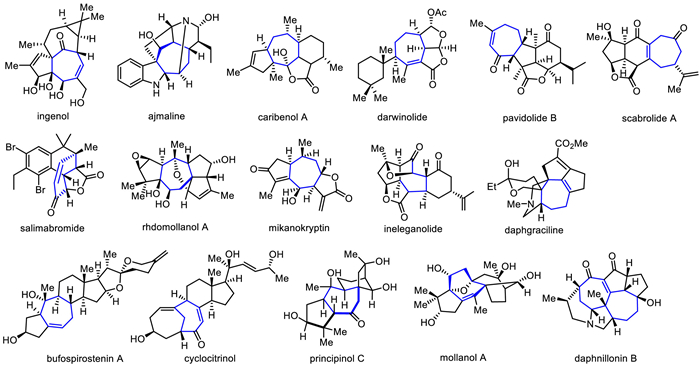
Bufospirostenin A, an unusual steroid with rearranged A/B rings, was isolated by Ye's group in 2017 from the toad Bufo bufo gargarizans [20]. It is a class of complex steroid natural products with a unique 5/7/6/5/5/6 cohesive hexacyclic skeleton and eleven chiral centers, including ten continuous chiral centers and two quaternary carbon centers.
Li's group accomplished the first and asymmetric total synthesis of bioactive bufospirostenin A [21] in 2020, featuring a unique intramolecular rhodium-catalyzed Pauson-Khand reaction of an alkoxyallene-yne as key step (Scheme 2). In their synthesis, the Pauson-Khand precursor 8 was converted to readily available enone 7 in five steps involving C-alkylation, 1,4-reduction, removal of the acetal protecting group, Seyferth-Gilbert homologation, and 1,2-addition with allene lithium reagent. The following intramolecular Pauson-Khand reaction was utilized to construct the seven-membered ring to give the desired [5-7-6-5] tetracyclic core 9 from 7, which was further converted to intermediate 10 through a nine-step sequence in 10% overall yield. Next, diastereoselective hydroboration-oxidation of the alkene and reduction of the ethyl in 10 to give diol, followed by DMP oxidation, Pinnick oxidation, esterification, chemoselective reduction of the ketone group, and a subsequent TBAF workup gave the anticipated lactone 11 in three steps. Interestingly, compound 11 underwent an elimination with SOCl2 and pyridine to generate the desired double bond, followed by treatment with lithium reagent generated from 12 with t-BuLi, a subsequent HCl workup for spiroketalization and deprotection gave bufospirostenin A.
Rearrangement reactions play a key role in organic synthesis, especially in the total synthesis of natural products, although the bond cleavage and formation in these reactions are not usually intuitive. Typically, a new and complex skeleton is constructed by an easily accessible precursor through a rearrangement reaction that includes an active intermediate, such as the carbon cation. Recently, this strategy have been successfully applied to afford seven-membered rings of natural product by the following groups, Gui [22–25], Yang [26], Ding [27,28], Baran [29], Qiu [30] and Stoltz [31].
Propindilactone G is a type of highly oxidized diterpenoid natural product with a unique 5/5/7/6/5-ring system and ten carbon chiral centers, including two quaternary chiral centers and one all-carbon quaternary chiral center, isolated from the stem vine of Schisandra propinqua var. propinqua by Sun's group in 2008 [32]. Preliminary studies on biological activity have shown that these compounds have potential anti-HIV activity. Their diversity, complexity, and potential biological properties have attracted great attention from synthetic chemists.
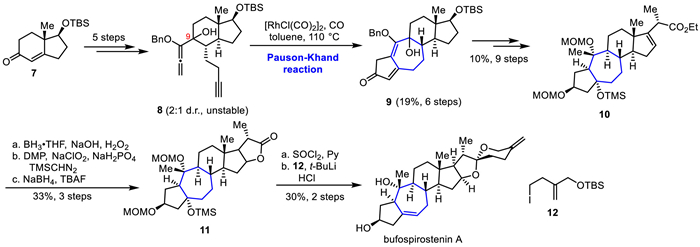
In 2020, Gui's group completed the total synthesis of propindilactone G using a biomimetic synthesis strategy inspired by the biosynthetic hypothesis proposed by Sun's group (Scheme 3) [22]. Their work commenced with the synthesis of 14, which was prepared efficiently from commercially available compound 13 in four steps. The following remote intramolecular radical cyclization and reduction with Zn were utilized to construct the C19-OH 15 in two steps. Using a one-pot method, the fused product 16 was obtained through a Wagner-Meerwein rearrangement reaction.
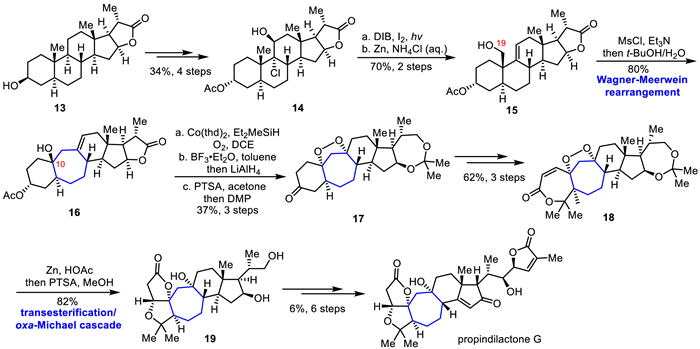
Subsequently, the Mukaiyama hydration and intramolecular cyclization reactions were applied to achieve the inversion of the tertiary hydroxyl stereo center at the C10 position, followed by several steps of functional group transformation affording ketone 17. The unsaturated lactone 18 was obtained by Baeyer-Villiger oxidation and Mukaiyama dihydroxylation in three steps. Under Zn/HOAc conditions, simultaneous peroxy bond reduction, ester exchange, and oxygen hetero-Michael cascade reaction were performed in situ to construct the 5/5-fused bicyclic lactone portion. The diol 19 was obtained after removing the allyl protection under PTSA. Finally, the butenolide ring of the propindilactone G was constructed in six steps involving Swern oxidation, HWE olefination, Saegusa oxidation, epimerization, and Sharpless asymmetric dihydroxylation.
In 2021, Gui's group proposed two possible biomimetic synthesis pathways, they designed that the 5/7-fused ring structure in the natural product was obtained by the Wagner-Meerwein rearrangement or the Photosantonin rearrangement of the 6/6-fused ring structure [23]. Subsequently, they developed two different biomimetic routes based on the Photosantonin rearrangement and Wagner-Meerwein rearrangement, respectively, furnishing the total synthesis of bufospirostenin A from the same starting material tigogenin lactone 13. The synthetic route based on the Wagner-Meerwein rearrangement was shown in Scheme 4a. Dehydrogenation reaction of the commercially available steroid lactone 13 proceeded smoothly in the presence of m-iodoxybenzoic acid and diphenyl diselenide to afford dienone 20, which was converted to compound 21 through three steps. The key precursor 22 was obtained by three simple functional group transformations from compound 21. The 5/7-fused ring 23 was successfully constructed by the Wagner-Meerwein rearrangement from compound 22. Finally, the asymmetric biomimetic synthesis of bufospirostenin A was completed according to the method reported by Li's group [21].

Meanwhile, Gui's group also achieved the synthesis of bufospirostenin A through the Photosantonin rearrangement strategy (Scheme 4b) [23]. Treatment of the same dienone intermediate 20 with iodination and palladium-catalyzed carbonyl insertion smoothly produced the rearrangement precursor 24. Under AcOH and light conditions, the Photosantonin rearrangement occurred, resulting in the highly fused lactone compound 25, which was followed by saponification, decarboxylation, acetylation, and Luche reduction, the allylic alcohol 26 was obtained in three steps. Subsequently, the double bond isomerization was completed by using Co-catalyzed hydrogen selenation and oxidation elimination reaction. Finally, based on the method reported by Li's group, the asymmetric biosynthesis of bufospirostenin A was completed [21].
In 2022, Yang's group reported the asymmetric biomimetic synthesis of bufospirostenin A using a Photosantonin rearrangement and a regioselective Co-catalyzed olefin isomerization strategy (Scheme 4c) [26]. The rearrangement precursor 29 was achieved through three steps of functional group transformations, which was prepared efficiently from the known dienone intermediate 20. Under the Photosantonin rearrangement reaction, the highly-fused 5/7 ring system 30 was smoothly produced in 54% yield, which underwent Krapcho reaction to give the decarboxylation product 32 (22%) and lactone 31 (71%) under NaCl conditions. Compound 31 was then treated with the action of strong base KOH affording 32 in 30% yield. Therefore, the overall yield for formation of 32 from 30 was 78%. Then, reduction of 32 followed by MOM protection gave 33 in 52% yield. With compound 33 in hand, the authors focused on the isomerization of the bridged double bond. Treatment of 33 with Co catalyst cat-1 and PhSiH3 successfully produced 34 through HAT process in 56% yield. Finally, the spiroketal part was assembled using a previously reported method to complete the synthesis of bufospirostenin A [21].
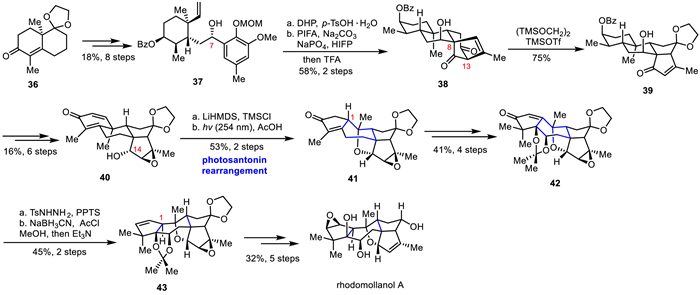
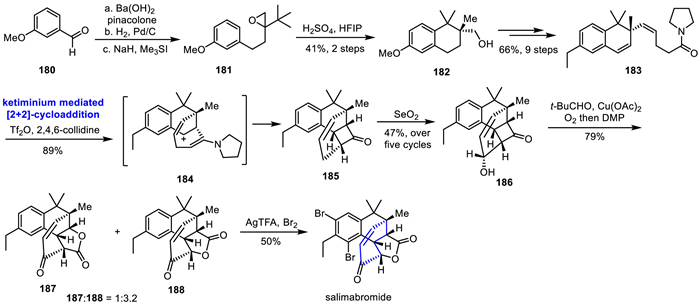
In 2017, Yao's group isolated a novel diterpenoid, rhodomollanol A, with a high oxidative state from the leaves of Rhododendron mole [33]. Rhodomollanol A has a unique 3/5/7/5/5/5 hexacyclic skeleton and 11 consecutive chiral centers, together with a rare 7-oxa-2-azabicyclo[4.2.1]nonane scaffold. Inspired by the biosynthesis of rhodomollanol A, Ding's group ingeniously designed three cascade reactions to efficiently construct the rhodomollanol A ring skeleton, and completed its asymmetric total synthesis through multi-step functional group transformations in 2020 (Scheme 5) [27]. The authors achieved the ideal intermediate 37 via eight-step sequence of transformation from Wieland-Miescher ketone derivative 36 in 18% yield. Treatment with DHP and TsOH, the C7 position was protected by THP and the MOM protection group was removed. Then, a single compound 38 was obtained through ODI-[5 + 2] cycloaddition/1,2-acyl migration cascade reaction. Subsequently, the reverse Dieckmann condensation and the enyne insertion Dieckmann cyclization cascade reaction of 38 to construct the [3.3.0] bicyclic structure 39 in one pot. Further transformations were carried out to complete the rearrangement precursor 40 in six steps. Selective protection of C14-OH followed by photosantonin rearrangement under light and AcOH, and due to the slow release of the C14 hydroxy group in the presence of acetic acid, an intramolecular etherification reaction occurred, resulting in the formation of compound 41 and completing the construction of the entire molecular framework. While the C1 stereocenter was inconsistent with the target molecule, they converted the unsaturated ketone in 42 to corresponding nitrile, obtaining compound 43. Finally, through stereoselective and region-selective oxidation and reduction, the first asymmetric total synthesis of the highly oxidized complex diterpene rhodomollanol A was achieved.
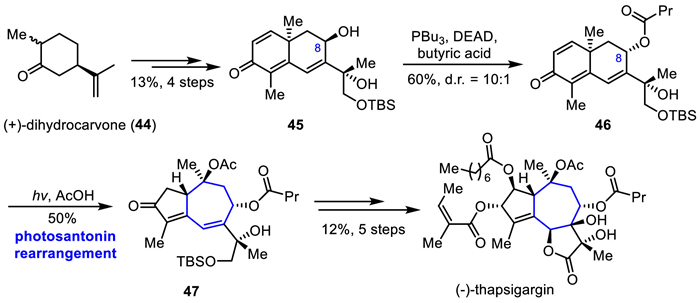
Thapsigargin is a complex and highly oxidized sesquiterpene lactone isolated from the Mediterranean plant Thapsia garganica by Christensen and co-workers in 1978 [34–36]. Thapsigargin is a highly potent inhibitor of intracellular calcium ion channels, with selectivity reaching the nanomolar level. Structurally, thapsigargin has a highly oxidized 5/7/5 tricyclic yuzurine lactone ring, together with eight carbon chiral centers, four different ester groups, one trans-fused bicyclic vicinal diol, and one tetrasubstituted olefin.
In 2017, Baran's group reported an elegant synthesis of (−)-thapsigargin in a scalable and more concise fashion by utilizing two-phase terpene [29]. As shown in Scheme 6, their work commenced with the synthesis of allylic alcohol 45, which was prepared efficiently from (+)-dihydrocarvone 44 over four steps. Mitsunobu inversion with butyrate acid allowed for the smooth installation of the butyrate with the desired stereochemical configuration at C8. Then the key photosantonin rearrangement was attempted. Treatment of 46 with a Hg lamp in glacial acetic acid furnished enone 47 in 50% yield, thereby stereoselectively constructing the guaianolide skeleton. Under the presence of octanoic acid/octanoic anhydride and KMnO4 heating conditions, the desired α-octanoylated enone was achieved. Subsequently, the pentacyclic ring of thapsigargin was constructed by applying dihydroxylation and Parikh−Doering oxidation reactions. Finally, the total synthesis of the complex highly oxidized sesquiterpene thapsigargin was completed via Zn(BH4)2 reduction and Yamaguchi esterification reaction. This constitutes a 11-steps synthesis of (−)-thapsigargin with 0.137% overall yield.
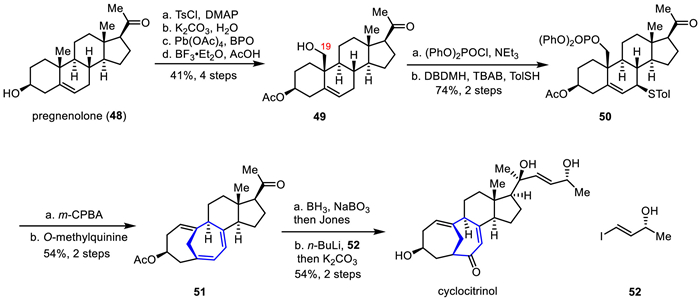
Cyclocitrinol was isolated in 2000 by Gräfe and co-workers from the secondary metabolites of the land citrus mold Penicillium citrinum [37]. Structurally, it contains a crowded 7/7/6/5 tetracyclic framework and a unique bicyclo [4.4.1] bridge system, with eight chiral centers, including two quaternary centers. In 2018, Gui's group reported a bioinspired strategy that has enabled a scalable synthesis of cyclocitrinol in only 10 steps from available material pregnenolone (48, $0.32/g) [24,25]. This key synthesis features a cyclopropane ring-opening rearrangement tandem reaction to concisely and efficiently construct the challenging a bicyclo [4.4.1] bridged ring system [38].
The synthetic route was shown in Scheme 7. Their work commenced with the synthesis of tetracyclic compound 49, which was prepared efficiently from commercially available pregnenolone 48 and selective oxidation of the C19 methyl group through four steps of functional group transformation in 41% yield. Subsequently, the rearrangement precursor 50 was obtained from compound 49 through phosphorylation, bromination, and SN2 substitution. Treatment of 50 with m-CPBA gave sulfone, followed by the biomimetic tandem rearrangement reaction using O-methylquinine as base to construct the bicyclo [4.4.1] bridge framework of 51 in two steps. Compound 51 was underwent hydroboration and followed oxidized by Jones reagent giving the dicarbonyl compound, which was then converted to the natural product cyclocitrinol via a tandem 1,2-addition and deacetylation.
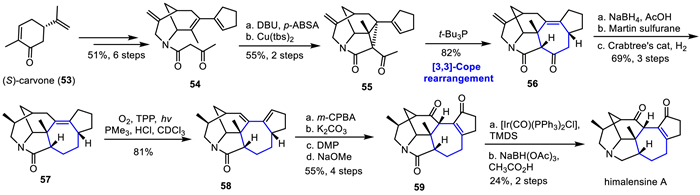
Himalensine A, a member of the calyciphylline A type of Daphniphyllum alkaloid, was isolated by Yue and co-workers from Daphniphyllum himalense in the Himalayan region in 2016 [39]. The himalensine A has a 2-azabicyclo-[3.3.1]nonane core structure bearing six stereogenic centers, which makes it a challenging synthetic target. In 2021, Qiu group accomplished the asymmetric total synthesis of himalensine A in 19 steps, featuring the [3,3]-Cope rearrangement as the key reaction to construct the himalensine A skeleton (Scheme 8) [30]. Their work commenced with synthesis of compound 54, which was prepared efficiently from (S)-carvone 53 over six steps in 51% yield. Next, diazotization of 54 followed by Cu(tbs)2-catalyzed cyclopropanation afforded the precursor 55, which underwent a Cope rearrangement using P(t-Bu)3 as the reagent to produce cycloheptenone 56 in 82% yield. Next, cycloheptenone 56 was converted to the core skeleton of himalensine A 57 over three steps, involving NaBH4 reduction/Martin sulfurane dehydration, and hydrogenation using Crabtree's catalyst. Allylic transpositional oxidation of 57 via Schenck-ene reaction proceeded smoothly with singlet oxygen using tetraphenylporphyrin (TPP) as the photosensitizer, then reduction of allylic peroxide using PMe3 followed by dehydration to generate diene 58 in 81% yield. Subsequent the regioselective epoxidation was accomplished under m-CPBA conditions, subsequent SN2′ ring opening by the in situ formed m-chlorobenzoic acid, followed by deprotection with K2CO3 to give the diol, then DMP oxidation followed by olefin isomerization under NaOMe as base furnished 59 in four steps. Finally, an Ir-catalyzed chemoselective reduction of the lactam carbonyl group complete the asymmetric total synthesis of himalensine A in 24% yield.
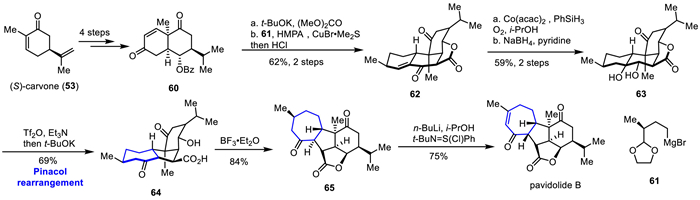
Pavidolide B is a novel diterpenoid marine natural product isolated from the soft coral Sinularia pavida by Lin and co-workers in 2012 [40]. In 2019, Ding's group achieved the asymmetric total synthesis of pavidolide B through two Mukaiyama Michael additions, ester exchange/Dieckmann condensation cascade reaction, pinacol rearrangement, and late-stage BF3·Et2O-mediated lactonization [28]. As shown in Scheme 9, they started with the (S)-carvone 53 and transformed it into 60 in four steps through the first Mukaiyama-Michael addition, followed by HF deprotection of the TBS group, the second Mukaiyama-Michael addition, and elimination, resulted in cis-[6,6] bicyclic diketone 60, which underwent ester exchange/Dieckmann condensation cascade reaction in the presence of t-BuOK and dimethyl carbonate to produce lactone. A one-pot synthesis using organic copper reagents mediated stereoselective Michael addition with 61 and intramolecular aldol condensation reaction resulted in the formation of tetracyclic compound 62. Using the Mukaiyama hydration reaction and NaBH4/pyridine reduction, cis-diol 63 was obtained in 59% yield over two steps. The secondary hydroxyl group in 63 was treated with Et3N and Tf2O to produce the corresponding triflate, followed by Pinacol rearrangement under alkaline conditions, yielding the bicyclic product 64 in 69% yield. Treatment of 64 with BF3·Et2O, the cyclization to [3,5,5,7] tetracyclic intermediate was followed by intramolecular nucleophilic addition of the carboxylic acid group, leading to the opening of the cyclopropane ring and the successful construction of the five-membered lactone ring in 65. Finally, the Mukaiyama dehydrogenation was used to selectively oxidize the C8 and C9 positions and isomerize the C11 position, completing the asymmetric total synthesis of pavidolide B.
Ineleganolide was isolated from the Formosan soft coral Sinularia inelegans by Duh and co-workers in 1999 [41]. It has shown preliminary cytotoxicity against P-380 leukeumia cell lines. Unlike the other family members, ineleganolide contains a highly rigid oxidized framework that bears a key central seven-membered ring, a remote isopropenyl group, and a bridging β-ketotetrahydrofuran moiety. These structural features render ineleganolide an even more challenging target for total synthesis over the past two decades. Only, in 2022, Wood's group accomplished the first total synthesis of ineleganolide using a transannular Michael reaction as the key strategy [42].
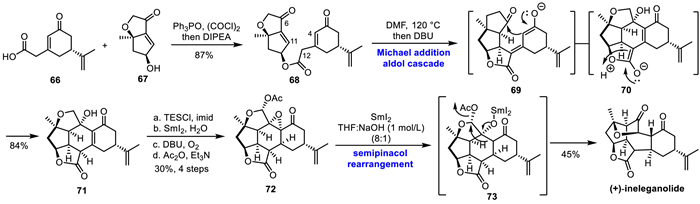
In 2023, Stoltz's group completed the convergent total synthesis of diterpenes (+)-ineleganolide derived from butenolides (Scheme 10) [31]. The key reactions of this total synthesis route include: (1) Esterification, linking two enantiomeric enriched fragments from (−)-linalool and (+)-norcarvone; (2) Michael addition/aldol addition tandem reaction, two bonds and four stereocenters are constructed in one step; (3) Allyl oxidation of conjugated ketenes and double bond epoxidation of olefin promoted by oxygen; (4) SmI2 mediated epoxyopen-ring/semi-pinacol rearrangement reaction to furnish the highly rigid central seven-membered ring. The synthesis commenced with the TPPO/(COCl)2-mediated esterification of compounds 66 and 67. Using (+)-norcarvone and (−)-linalool as starting materials, compounds 66 and 67 were synthesized and esterified to produce ester 68 in 87% yield after the author carried out the screening of esterification conditions. While they evaluating the Michael addition conditions, ester 68 was unstable to various bases, but under the promotion of base DBU, an unexpected tandem reaction of Michael addition/Aldol cyclization occurred, and the final product 71 (X-ray) was generated through intermediates 69 and 70, and two bonds and four stereocenters were constructed in one step. Treatment of 68 in DMF at high temperature and adding DBU in one portion to smoothly give 71 in 84% yield within minutes. Next, the pentacycle 71 was converted to acetate 72 over four steps in 30% yield, which followed by a reduction epoxy ring-opening reaction promoted by samarium diiodide, and semi-pinacol rearrangement via intermediate 73 to produce the natural product (+)-ineleganolide.
The radical is a very important intermediate to form the new rings. Due to its high reactivity, the radical involved reactions have not received much attention for many years. In the development of EPR, the radical pathway emerged as a powerful tool to build complex skeletons. In particular, the C−C bond formation reactions via radical pathway is particularly useful and has been applied in the total synthesis of natural products successfully in recent years [43–46].
Like other grayanane-type diterpenoids, principinols have a highly oxidized tetracyclic skeleton, as well as multiple chiral centers and a bicyclic [3.2.1] octane structure. In 2019, Newhouse's group used a convergent synthetic strategy to assemble the core skeleton by assembling two chiral fragments through a late-stage coupling [47]. As shown in Scheme 11, they started from cyclohexenone 74 and after four steps of transformation, obtained enyl bromide 75. In the presence of NiCl2(PCy3), a cross-coupling reaction between the intramolecular enol salt and the enyl bromide successfully constructed the bicyclic [3.2.1] octane structure, and in situ deprotection of TBS with HCl led to the formation of ketone 76. Subsequently, iodide 77 was obtained through SmI2 reduction, Appel reaction and MOM protection. After five steps of transformation, the cyclization precursor 79 was obtained in 15% yield. Under the presence of water, the SmI2-mediated reduction-coupling reaction of carbonyl-alkene was carried out to obtain 80, thus completing the construction of the entire molecular skeleton. Finally, after five steps of functional group transformation, the first total synthesis of principinol D has been completed.

Resiniferatoxin belong to a daphnane diterpenoid family, which was as an irritant component isolated from the latex of Euphorbia resinifera by Hecker and co-workers in 1975 [48]. Initial biological activity testing showed that resiniferatoxin has strong agonistic effects on TRPV1, a transducer of noxious temperature and chemical stimuli [49]. And the molecule structure elucidation in 1982 revealed, which is characterized by a densely oxygenated trans-fused 5/7/6-tricarbocycle, a C9,13,14-orthoester and a C13 isopropenyl group, so it made its synthesis highly challenging for chemist [50].
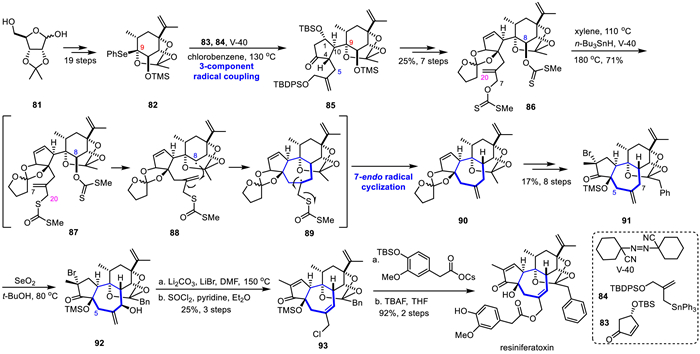
In 2017, Inoue's group completed the total synthesis of resiniferatoxin using a convergent synthesis strategy, including key reactions such as intermolecular 3-component radical coupling and intramolecular 7-endo radical cyclization, providing a basis for further structural modification and structure-activity relationship research (Scheme 12) [51]. The pivotal intermediate 82 of the radical-based strategy was constructed from the commercially available D-ribose derivative 81 in 19 steps. 3-Component radical coupling of 82 with 83 and 84 in the presence of V-40 produced 85 efficiently as a single isomer. This first radical coupling generated the hindered C9,10-linkage between the 5-membered and 6-membered rings and extended the C4-chain in a stereoselective fashion. After derivatization of 85 into the C7- and C8- of bisxanthate 86 in seven steps, the radical cyclization precursor 86 was obtain in 25% yield. Under 110 ℃ in xylene, [3.3]-sigmatropic rearrangement of 86 into dithiocarbonate of 87. Then, the tricycle 79 was achieved as the only isolable product in 71% yield under microwave irradiation in the presence of n-Bu3SnH and V-40. The second radical reaction thus enable not only the 7-endo cyclization but also formation of the C8-stereocenter and C6-exo olefin. The tricycle 90 was then converted to C2-brominated 91 in eight-steps transformations. Although 91 has the three potential reactive sites (C5, 7, 16), allylic hydroxylation of 91 in the presence of SeO2 in t-BuOH to generate the C7-alcohol 92 as the major product, which was followed by elimination of HBr and chlorination with SOCl2 to afford the chloride 93 in 25% overall yield. Finally, the SN2 reaction of 93 with cesium carboxylate, followed by deprotection with TBAF gave the targeted resiniferatoxin.
In 2020, Trost's group developed a Pd-catalyzed cycloisomerization reaction of polyenyl acetylenes, followed by intramolecular acyl radical cyclization to complete the total synthesis of the sesquiterpenoid natural product 10,11,12-trihydroxytremulenone [52]. As shown in Scheme 13, they started from commercially available propargyl alcohol and obtained a large amount of diketone 94 through one-step transformation. The single methyl ester was obtained using n-BuLi and methyl chloroformate. Then, 1,2-addition of a terminal alkyne to a highly hindered fatty aldehyde 95 was performed using (S,S)-ProPhenol and ZnMe2 to obtain 96. Under the action of Pd2(dba)3·CHCl3/P(o-tol)3, the enyne undergoes a cycloisomerization reaction to afford the cycloisomerization intermediate 97, which was treated with a catalytic amount of TsOH to undergo a tandem transformation to obtain compound 98. Subsequently, 1,4-reduction, saponification, and selenylation were performed to produce acyl selenide 99. Under the conditions of Et3B/air as an initiator and (TMS)3SiH as a hydrogen source, 7-exo radical cyclization occurs, constructing the 5/7 fused ring system 100. Finally, after further processing, the asymmetric total synthesis of 10,11,12-trihydroxytremulenone has been completed.

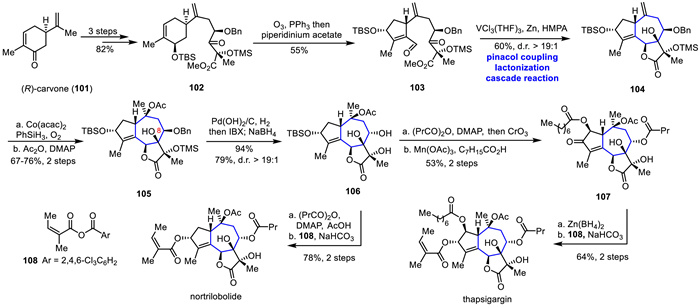
In 2017, Evans's group based on a convergent synthetic strategy, constructed the key tricyclic intermediate through a vanadium-mediated intramolecular pinacol coupling/internal esterification cascade reaction, and then efficiently completed the total synthesis of thapsigargin and nortrilobolide through a series of functional group transformations (Scheme 14) [53]. (R)-carvone 101 was converted to 102 in three steps. Selective ozonolysis of the trisubstituted double bond on the six-membered ring followed by intramolecular aldol reaction produced the unsaturated aldehyde 103. Under the action of the in situ formed dinuclear vanadium complex [V2Cl3(THF)6]2[Zn2Cl6], 103 underwent pinacol coupling and in situ ester exchange to afford the coupled product 104, completing the construction of the entire molecular skeleton. Then, the 104 underwent Mukaiyama hydration with Co(acac)2 in the presence of the PhSiH3 and O2 followed by acetyl protection to provide 105. Through a one-pot hydrogenation, oxidation, and reduction reaction, the C8 hydroxy group in 105 was stereochemically inverted to obtain 106. Subsequently, esterification and Jones oxidation were carried out to obtain the enone. The hydroxyl group at the α-position of the carbonyl group was oxidized with stereochemical selectivity under trivalent manganese conditions, and the esterification was carried out in situ to obtain 107. Finally, through reduction and Yamaguchi esterification reaction, the total synthesis of thapsigargin was completed. The entire synthetic route was concise and efficient, and could be carried out on a kilogram scale. In addition, starting from the common intermediate 106, the total synthesis of the target natural product nortrilobolide was completed through esterification, TBS removal, and Yamaguchi esterification reactions.
In 2022, Sarpong's group has completed the total synthesis of nine oxygen-containing Longiborneol family natural products using a concise and efficient strategy based on semi-pinacol rearrangement and metal-hydride hydrogen atom transfer (MHAT) radical cyclization (Scheme 15) [54]. Commercially available (S)-carvone 53 was subjected to m-CPBA epoxidation and Ti(Ⅲ)-mediated reduction coupling to give the diol 109 in 76% yield (d.r. = 1.5:1 at C7). Then, treatment of 109 with p-TsOH acid resulted in the semi-pinacol rearrangement reaction to produce 8-hydroxyborneol 110, which was further oxidized to aldehyde under TMEPO and PIDA conditions. The dienone intermediate was provided through the wittig reaction, which contains all the carbon atoms of the longiborneol class sesquiterpenes. Considering the metal-mediated MHAT radical cyclization reaction, a polar match is required between the radical donor and acceptor. They screened various enol derivatives of dienone and found that using vinyl phenyl sulfonate 111, the HAT radical cyclization reaction could be successfully carried out under Fe(acac)3 and PhSiH3 conditions, and the key intermediate 112 was obtained with 86% yield. Finally, through hydrogenation and Li/NH3(l) reduction, the asymmetric total synthesis of longiborneol was achieved in a concise and efficient manner in 9 steps with 30% overall yield. Meanwhile, they also completed the total synthesis of eight other oxygen-containing longiborneol sesquiterpenes from the advanced intermediate 112 and natural longiborneol through late-stage C−H functionalization.

Barbier reaction utilized the organic halides with a metal(0) to form the organometallic reagent in-situ, which then reacted with the carbonyl compounds (aldehydes or ketones) to obtain the corresponding secondary or tertiary alcohols. Due to the flexibility and high stereo-selectivity of the reaction, the allylation of carbonyls has been widely used in organic synthesis, which were carried out by reacting the target carbonyl group with the alkyl halide and a metal powder or reacting with a pre-formed allylic metal (or pseudo-metallic boron and silicon) reagent [55,56].
In 2017, Maimone's group completed the synthesis of mikanokryptin using a metal-mediated Barbier-type double allylation strategy [57]. As shown in Scheme 16, the chiral (S)-carvone 53 underwent chlorination under sulfonyl chloride, followed by Luche reduction, TBDPS protection. Subsequently, the selective oxidation of the trisubstituted double bond on the six-membered ring was carried out under pyridine conditions, followed by an aldol reaction under piperidine/acetic acid to obtain unsaturated aldehyde 113. Subsequently, the first Barbier-type allylation reaction with bromide 114 under metal indium gave compound 115 with a diastereoselective ratio of 2:1. Notably, one equivalent of water was added during the process to inhibit lactonization. The TES protection and removal of the acetal protection group under TESOTf and collidine conditions gave aldehyde 116. The intramolecular Barbier allylation reaction and in situ lactonization were carried out under SnCl2 and NaI conditions to obtain tricyclic compound 117, completing the entire molecular skeleton. The synthetic route was then adjusted to utilize Michael addition catalyzed by sodium methoxide, followed PtO2-catalyzed hydrogenation to successfully reduce the exocyclic double bond on the seven-membered ring and obtain compound 118. Then, TBAF removed the two silicon protection groups in 118, and β-elimination occurred with DBU to reform the exocyclic double bond of the five-membered lactone ring. Finally, selective oxidation of the allylic hydroxyl group on the five-membered ring was carried out under MnO2 condition, completing the first total synthesis of the natural product mikanokryptin.

Friedel-Crafts reaction is a powerful and classical tool to produce the substituted aromatic compounds in the presence of Lewis acids or Brønsted acids [58]. Intramolecular Friedel-Crafts reactions to construct the desired ring with the following advantages, inculding excellent reactivity, the diversity of the precursor compounds, and the predictability of the desired product. In recent years, this strategy has been widely used in total synthesis of natural products.
Caribenol A is a novel diterpenoid natural product isolated from Pseudopterogorgia elisabethae in 2007 by Rodríguez's group [59]. Structurally, caribenol A features a 5/7/6 tricyclic carbon skeleton with six stereocenters. The molecule has a cage-like structure and poses a great challenge in synthesis. In 2017, Trauner's group completed the total synthesis of caribenol A through Friedel-Crafts reaction to construct the seven-membered ring and subsequent furan epoxidation as key steps (Scheme 17) [60]. They started from the known furan derivative 119 and performed 10 steps transformations to obtain 120 in 25% yield. The attacking of Weinreb amide in 120 by vinyl Grignard reagent formed an unsaturated ketone intermediate, which was then cyclized through RCM to give the cyclopentenone 121. Under the action of 2,6-di-tert-butylpyridine and Tf2O, an intramolecular Friedel-Crafts alkylation reaction was forged, successfully constructing the seven-membered ring and producing the bowl-shaped tetracyclic compound 122. Treatment with TMSOK gave the ketone 123. Oxidation of the furan ring in 123 with peroxyacetic acid produced the butenolide 124. Finally, using the Barton method, the ketone carbonyl in 124 was converted to the vinyl iodide, followed by Stille coupling to complete the total synthesis of caribenol A.

Ring-closing metathesis (RCM) reaction is alternatively utilized to construct cyclic medium-sized and larger rings. In addition, the heterocycles can also be obtained using this method [61–65]. Recently, RCM reaction has become a key strategy for the total synthesis of natural products containing seven-membered carbon rings.
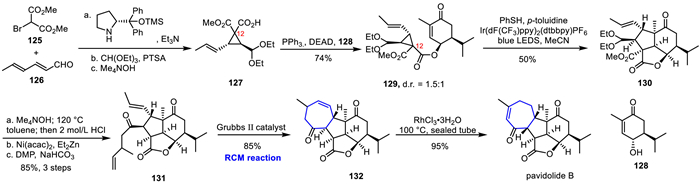
In 2017, Yang's group completed the first asymmetric total synthesis of the marine natural product pavidolide B using key strategies such as visible light-mediated sulfur radical cascade cyclization and RCM reaction [66]. As shown in Scheme 18, bromoester 125 reacted with unsaturated aldehyde 126 via a chiral pyrrole catalyst to carry out Michael/alkylation cascade reaction to obtain vinyl cyclopropane. Subsequently, aldehyde protection and selectively saponified was carried out, producing the cyclopropane 127. The Mitsunobu reaction of 127 with 128 obtained the radical cyclization precursor 129 in 74% yield. Then, after the extensively screening of the key radical cascade cyclization conditions, 130 was obtained stereoselectively in 50% yield at room temperature under blue light irradiation with [Ir(dF(CF3)ppy)2(dtbbpy)PF6] as photosensitizer, acetonitrile as solvent, and thiophenol as reagent. After saponification, decarboxylation, and removal of the acetal protecting group, an aldehyde has been achieved, which underwent allene propargylation reaction, followed by DMP oxidation to obtain diene 131. RCM cyclization reaction was carried out using Grubbs Ⅱ catalyst to complete the construction of the entire molecular skeleton, obtaining compound 132. Finally, catalytic amount of RhCl3·3H2O was used under sealed heating conditions to induce the migration of the double bond, and at the same time, the α-position of the seven-membered ring ketone carbonyl underwent a difference-directed isomerization, completing the first asymmetric total synthesis of pavidolide B.
Darwinolide is a rearranged sesterterpenoid isolated by Baker's group from the Antarctic sponge Dendrilla membranosa in 2016 [67]. It exhibits potent inhibitory activity against the biofilms of methicillin-resistant S. aureus (MRSA) and is a potential candidate for a new type of antibiotic targeting biofilms specifically. Structurally, darwinolide features a [3.3.0]-dioxabicyclooctanone strained fused to a cycloheptene ring, which is connected to a cyclohexyl unit and a tetrasubstituted double bond. It is believed to be produced by cyclization and rearrangement of a common spongian precursor.
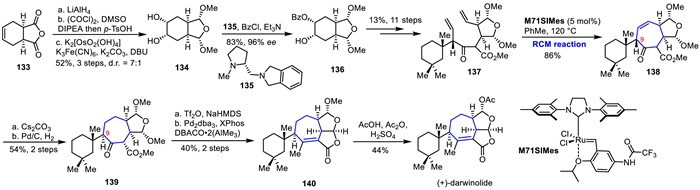
In 2019, Christmann's group achieved the first asymmetric total synthesis of (+)-darwinolide by using a convergent aldol fragment coupling, an organocatalytic desymmetrization and RCM reaction as key reactions for constructing the skeleton of (+)-darwinolide (Scheme 19) [68]. They used commercially available anhydride 133 as the starting material, which was reduced by LiAlH4, oxidized by Swern oxidation, and underwent acid-catalyzed double aldolization and Sharpless dihydroxylation to obtain diol 134. Under the action of a chiral amine catalyst, desymmetrization of meso-diol 134 was accomplished to give mono benzoyl ester 136 with excellent enantioselectivity (96% ee). Subsequently, after 11 steps of functional group transformation, RCM precursor 137 was obtained in 13% overall yield. The RCM reaction was successfully carried out to construct the core heptacyclic structure 138 using Umicore's M71SIMes catalyst in 86% yield. Epimerization at C9 position of 138 was accomplished under basic conditions to produce the thermodynamically favored β-keto ester, followed by hydrogenation reduction to afford 139 in 54% yield over two steps. Next, the authors converted β-keto ester 139 into the corresponding vinyl triflate, and using Pd2dba3 and XPhos as precatalysts, DABCO and Me3Al in a 1:2 ratio as a methyl source, they carried out Woodward's coupling reaction, simultaneously constructing the unstable lactone hemiacetal and tetrasubstituted olefin, to give compound 140, followed by acetylation, accomplished the first asymmetric total synthesis of (+)-darwinolide.
The oxidopyrylium ylide [5 + 2] cycloaddition reaction plays an important role to produce seven-membered rings and has been used extensively in the synthesis of natural complexes. In 2014, Li's group developed a unique type Ⅱ [5 + 2] cycloaddition reaction to provide a facile and direct method to access the highly functionalized bridged bicyclo[m.n.1] ring motifs with a strained bridgehead double bond [69–71].
Cyclocitrinol, an unusual C25 steroid, was isolated from terrestrial Penicillium citrinum, and its structure was revised by Crews' group in 2003 [72]. Structurally, cyclocitrinol possess a sterically compact 7/7/6/5 tetracyclic which features a unique bridged bicyclo [4.4.1] A/B ring with two quaternary centers. Therefore, cyclocitrinol presents a formidable synthetic challenge. In 2018, Li's group accomplished the first asymmetric total synthesis of cyclocitrinol, and the key step is a type Ⅱ [5 + 2] cycloaddition to construct the strained bridgehead bicyclo [4.4.1] A/B ring system [73]. The synthesis was initiated with the preparation of 142 (Scheme 20), which was achieved from 141 in 31% yield over five steps. Treatment of 142 with lithium reagent generated from bromide 143 and removal of both silyl groups, followed by oxidation rearrangement in situ by NBS, generated 144. Then, the key type Ⅱ [5 + 2] cycloaddition occurred using TMP as a base with heating, to obtain 7/7/6/5 tetracyclic ring system 145 in four steps. The carbonyl group of 145 was further removed to afford 146 in three steps, next challenging task was the site-selective cleavage of the allylic C18-O. After some failed conditions were screened, treatment of the tertiary alcohol 146 with SOCl2, pyridine and colidine followed by using lithium metal in EtNH2 gave 147. Chemoselective oxidation of two primary hydroxyl group in 147 afforded the desired dialdehyde, followed by TES protection. Pleasingly, the desired oxidative deformylation of dialdehyde using t-BuOK and O2 in t-BuOH afforded diketone, which was transformed to cyclocitrinol with lithium reagent generated from 148 and TBAF workup, completing the first total synthesis of cyclocitrinol in 18 steps.

The natural product Daphgraciline is one of the representative molecules of the Yuzurine-type alkaloid family, which was first isolated from the bark of Daphniphyllum gracile Gage by Yamamura's group in 1980 [74]. However, the total synthesis of any member of the Yuzurine-type subfamily has not yet been reported in the last 50 years. Structurally, it contains a spatially crowded [6-7-5-5-6] pentacyclic skeleton, as well as a unique bridged azabicyclo [4.3.1] ring system and a spiro tetrahydropyran moiety, making it extremely challenging to access. In 2022, Li's group has realized the first complete synthesis of Yuzurine alkaloid Daphgraciline by using a novel [5 + 2] cycloaddition and an intramolecular Diels-Alder reaction as the key strategy. The synthesis work has laid a solid foundation for the synthesis of other Yuzurine alkaloids in the future [75]. As shown in Scheme 21, this synthesis work started with the preparation of 151 from 149 in four steps. The desired type Ⅱ [5 + 2] cycloaddition of 151 was achieved by using DHQD as a base, providing 152 as a single diastereomer in 75% yield. It is the first example of type Ⅱ [5 + 2] cycloaddition proceeding at room temperature with DBU as the base. Next, diastereoselective 1,2-addition of 152 with 153 followed by IMDA reaction proceeded smoothly to give 154 as a mixture of C15-diastereoisomers (d.r. 2.3:1) in 72% yield. The mixture of C15-diastereoisomers of 154 were desired products, which were further converted to the diketone, followed by a benzylic acid-type rearrangement to give the compact 6/7/5/5 tetracyclic skeleton of 155 through a four-step sequence in 45% yield. The anticipated epoxide 156 was synthesized from 155 via a six-step transformation in 24% yield, after a short screening on generation of a new all-carbon quaternary stereocenter C5, they found that Ti(Ⅲ)-mediated reductive epoxide cyclization of 156 with acrylonitrile afforded the spirolactone 157 in 81% yield as the only diastereomer, which was converted to ester 158 in four steps. Subsequent a Schenck ene reaction of 158 using TPP as the photosensitizer followed by elimination with MgSO4 at 140 ℃ afforded the desired dehydrodaphgraciline, which was further converted to daphgraciline through hydration with p-TsOH in THF/H2O in 46% yield over three steps.

Steroid bufogargarizins A and B are natural steroid molecules isolated and identified from the venom of Bufo sinensis in 2010 by Ye's group [76]. In 2023, Li's group completed the first asymmetric total synthesis of bufogargarizin A using the first intramolecular Ru-catalyzed [5 + 2] cycloaddition to efficiently construct the 7/5/6/5 tetracyclic skeleton through a series of functional group transformations [77]. At the same time, through the novel retroaldol/transcyclic aldol tandem reaction, the recombination transformation of the 7/5/6/5 ring system to the 5/7/6/5 ring system was achieved, and the first asymmetric total synthesis of bufogargarizin B was completed. The synthesis began with the synthesis of compound 160, which was prepared efficiently from commercially available sitolactone 159 in nine steps (Scheme 22). Treatment of 160 with TMSOTf and DIPEA in DCE successfully produced the enol silyl ether 161 (Z/E = 2.5:1) in high yield, which was converted into the desired 7/5/6/5 tetracyclic core 162 in 81% yield from 160 by using the intramolecular [5 + 2] cycloaddition with [CpRu(CH3CN)3PF6] as a catalyst and NaHCO3 as a base, at 55 ℃ in DCE. Mukaiyama hydration of 162 gave 163 in 64% yield and the configuration of the enol silyl ether in 161 does not affect its reactivity and stereoselectivity. With the successful construction of the natural product backbone, the total synthesis of bufogargarizin A in 17 steps of chemical transformations. After screening several different conditions, compound 164 was a better substrate for the desired retro-aldol/transannular aldol cascade reaction. Treatment of 163 in four steps afforded 164 in 47% yield, followed by using DBU as a base in refluxing THF, producing compound 165 as a single diastereoisomer in 73% yield, which was further converted to 166 in two steps including DIBAL−H reduction, removal TBS group and DMP oxidation. According to a similar route to bufogargarizin A, the total synthesis of bufogargarizin B was achieved from 163.

Daphnillonin B (DaNB) was first isolated from the leaves and shoots of Daphniphyllum longeracemosa by Yue's group in 2019, which features a novel 7/6/5/7/5/5 hexacyclic core with a unique [4.3.1] bridge ring skeleton containing two seven-membered rings [78]. In 2023, Li's group successfully achieved the first complete synthesis of (±)- and (−)-DANB using cheap and easily available o-cresol as starting material. The key strategies include: Mild type Ⅰ [5 + 2] cycloaddition reaction, Pauson-Khand reaction, bionic Wagner-Meerwein rearrangement reaction [79]. Their work commenced with the synthesis of compound 168, which was prepared efficiently from o-cresol 167 in three steps (Scheme 23). Treatment of 168 with 169 successfully produced 170 through a tandem Mitsunobu reaction and deprotection. The Achmatowicz rearrangement of 170 with NBS, followed by workup with 2 mol/L HCl, afforded 171 in 77% yield, which was converted to 172 in one pot by a type Ⅰ [5 + 2] cycloaddition under very mild conditions at 25 ℃ in 77% yield. Then, 1,4-addition of Grignard reagent 173 to 172, followed by deprotection of Ns group, subsequent trichloroacetylation and acetylation, produced 174. The radical cyclization of 174 and dichlorination proceeded successfully in one pot with Grubbs Ⅱ and AIBN to form core scaffold 175, which was converted into 176 in three steps. Following a series of screenings on the Pauson-Khand reaction parameters, the optimal conditions was achieved to use Co2(CO)8 in refluxing MeCN, followed by iodination with ICl. The expected cycloaddition reaction proceeded in highly efficient and stereoselective forms to build the hexacyclic core with several desired stereocenters. In the following steps, in order to produce the 7/6/5/7/5/5 hexacyclic core by biomimetic rearrangement from the 6/6/5/7/5/5 hexacyclic framework, the authors successfully obtained the propose precursor 178 for the rearrangement from 177 in five steps. A Wagner-Meerwein-type rearrangement of 177 proceeded in the presence of o-dichlorobenzene (o-DCB) at 180 ℃ to produce the desired 7/6/5/7/5/5 hexacyclic core 179, followed by eight steps transformations to complete the total synthesis of daphnillonin B.

Salimabromide is a polyketide compound that was isolated in trace amounts (0.5 mg from 64 L of culture) from the marine actinomycete Enhygromxya salina by König's group in 2013 [80]. In 2018, Magauer's group completed the first total synthesis of salimabromide using Wagner-Meerwein rearrangement/Friedel-Crafts cascade reaction, enone imine ion intermediate [2 + 2] cycloaddition, and Baeyer-Villiger oxidation as key steps [81].
As shown in Scheme 24, starting from commercially available 2-methoxybenzaldehyde 180, the synthesis proceeded through Claisen-Schmidt condensation, hydrogenation, and Corey-Chaykovsky reaction to afford epoxide 181. Under catalytic concentrated sulfuric acid, 181 underwent intramolecular Wagner-Meerwein rearrangement/Friedel-Crafts cascade reaction to yield compound 182, using HFIP as a strong hydrogen-bond donor and a weak affinity. 183 was obtained in 66% yield through nine steps transformations. In the presence of 2,4,6-collidine and Tf2O, enone imine ion 184 underwent intramolecular [2 + 2] cycloaddition to construct a seven-membered ring and a rigid tetracyclic carbon framework, resulting in compound 185. Subsequently, under SeO2 conditions, the 185 underwent oxidation at the allylic position, with only a small fraction being converted to alcohol 186. Extending the time did not improve the yield. After performing the above process five times, 250 mg of compound 186 was obtained. Then, under Cu(OAc)2/O2 conditions, Baeyer-Villiger oxidation was carried out, followed by in situ DMP oxidation, yielding lactone 187 and 188 (d.r. 1:3.2) in one pot. Using a series of brominating reagents, only trace amounts of products were observed. Finally, they found that under AgTFA and liquid bromine conditions, bromination could be carried out to obtain the desired dibromo product salimabromide in 50% yield, completing the first total synthesis of salimabromide.
Scabrolide A is a norcembranoid-type marine diterpenoid natural product isolated by Sheu's group from the soft coral Sinularia scabra in 2002 [82]. In 2020, Stoltz's group utilized a convergent synthesis strategy to complete the first total synthesis of the diterpenoid natural product scabrolide A through intramolecular Diels-Alder reaction, enone-alkene [2 + 2] cycloaddition, and later ring-opening alcohol oxidation as key steps (Scheme 25) [83]. The allylic alcohol 189 and alkynyl fragment 190 were connected through a Steglich esterification reaction to obtain the ester. Subsequently, an intramolecular Diels-Alder reaction occurred in xylene solvent at 140 ℃, yielding a single cycloaddition product 191 in 85% yield. Under the action of VO(acac)2, a C8 tertiary hydroxyl-directed epoxidation occurred, yielding the epoxide. Under CpTiCl2 and Mn0 conditions, selective ring-opening of the epoxide yielded the diol, which was then oxidized with an IBX oxidant, resulting in a double bond migration and formation of the enone 192. Epoxidation of 192 with m-CPBA, followed by a ruthenium-catalyzed hydrosilylation of the alkyne to yield [2 + 2] cycloaddition precursor 193. Under 350 nm light, an intramolecular [2 + 2] cycloaddition reaction occurred, yielding a fused [6,4,5] ring system, which was then reduced by CpTiCl2 and Mn0 to obtain 194. The alkene was obtained through mercury-mediated Tamao-Fleming oxidation and Grieco dehydration. Finally, under CuI/NIS conditions, a ring-opening alcohol oxidation successfully constructed a seven-membered carbon ring, completing the first total synthesis of scabrolide A.

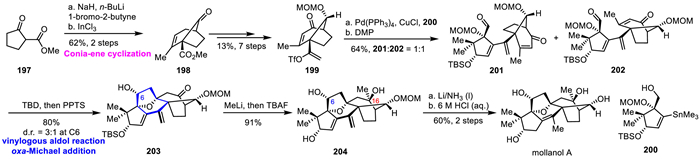
Mollanol A was isolated from the fruits of Rhododendron mole by Yu's group in 2014, which is the first member of the mollane-type grayanoids [84]. In 2022, Yang's group achieved the first total synthesis of mollanol A in 15 steps synthesis, featuring an InCl3-catalyzed Conia-ene cyclization and a vinylogous aldol reaction/intramolecular oxa-Michael addition as key steps (Scheme 26) [85]. Propargylation of commercially available 197 with 1-bromo-2-butyne, following Conia-ene cyclization to complete the bicyclo[3.2.1]alkenone 198 by using catalytic InCl3 in refluxing 1,2-dichloroethane (DCE). Subsequent the racemic C/D-ring fragment 199 was synthesized from 198 via a seven-step transformation, setting the stage for the cross-coupling with the A-ring fragment 200, which was obtained via a nine-step sequence from 2-methyl-1,3-cyclopentanedione (95% ee). The stille coupling of 199 and 200 in the presence of Pd(PPh3)4/CuCl efficiently produced a mixture of inseparable diastereomers, followed by DMP oxidation to afford 201 and 202 as a separable mixture (d.r. 1:1) in 64% yield. With tricyclic skeleton 202 in hand, after testing the vinylogous aldol reaction/oxa-Michael reaction condition to construct the key oxa-bicyclo[3.2.1] core, authors found that treating 202 with TBD gave the desired vinylogous aldol reaction, followed by the selective deprotection of MOM group, and the intramolecular oxa-Michael addition, providing 203 as a mixture of diastereomers (d.r. 3:1 at C6) in 80% yield. Treatment of 203 with methyllithium, followed by the removal of TBS group, to afford the 204 as a single diastereomer in 91% yield. 1,4-Hydrogenation of conjugated diene 204 using lithium and liquid ammonium as reductant, followed by removal of MOM group with HCl in methanol to achieve the mollanol A.
In 2022, Luo's group reported the total synthesis of (−)-grayanotoxin Ⅲ, (+)-principinol E, and (−)-rhodomollein XX based on a convergent strategy by using a Mukaiyama aldol reaction and a 7-endo-trig cyclization/1,2-migration as key reactions for constructing the skeleton of grayanane diterpenoids (Scheme 27) [86]. The synthesis of (−)-grayanotoxin Ⅲ commenced with a Vilsmeier reaction of 205 that was prepared via the asymmetric CBS reduction. The formation of dimethyl acetal proceeded smoothly to afford 206, which was treated with 207 gave 208 in 58% yield via a tandem reaction by using the organocatalytic Mukaiyama aldol and intramolecular Hosomi-Sakurai reactions in one pot fashion. The Suzuki coupling with potassium vinyltrifluoroborate followed by the treatment with NaOH afforded diol 209 in one pot with high yield, which was converted into 211 with 5/7/6/5 tetracyclic framework in two steps. The critical C7−C8 was forged to construct the seven-membered-ring by the highly reactive bridgehead carbocation via 7-endo-trig cyclization of a pendant alkene, while the process was terminated by deprotonation of 210 given the C2 proton was more acidic. Next, 212 was synthesized from 211 through a five-step transformation, which reacted with methyllithium, followed by epoxidation, DBU treatment, and TBS deprotection to afford enone 213 in 57% yield. The Mukaiyama hydration of 213 of C15−C16 olefin with aqueous H2SO4 solution to forge the tertiary alcohol in C16, which was further DIBAL−H 1,2-reduction, followed by the Mn(dpm)3 catalytic hydrogenation to give natural product (−)-grayanotoxin Ⅲ. Meanwhile, (+)-principinol E, and (−)-rhodomollein XX also were successfully accomplished by the late-stage intermediates en route as branching point.

In this review, we have summarized novel strategies towards the construction of the non-aromatic cycloheptane skeletons from 2017 to 2023, with emphasis on introducing the seven-membered ring motifs. Natural products containing a non-aromatic cycloheptane skeleton exhibit the promising biological activity, capturing the interest of synthetic and pharmaceutical chemists. Despite the efficient and elegant total synthesis of certain targets has been achieved, there are still many families of complex natural products have not yet been successfully synthesized. Furthermore, to address this challenge, there is a need for both improved existing methods and the development of novel unified strategies. This review encompasses a range of efficient methods, inculding Pauson-Khand reaction, rearrangement reactions, radical cyclization reactions, Barbier reaction, Friedel-Crafts reaction, ring-closing metathesis (RCM) reactions and intramolecular [5 + 2] cycloaddition, and their cascade transformations. Therefore, the pursuit of highly oxidative-reductive, atom-economic, and step-economical methods and strategies offers a novel approach to the asymmetric synthesis of medium-sized natural products and their derivatives, providing a solid foundation for further biological evaluations.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the financial support from the Start-up Grant of Henan University of Technology (No. 0004/31401540). T.-P. Loh thank the financial support from Distinguished University Professor Grant (Nanyang Technological University), AcRF Tier 1 grants from the Ministry of Education of Singapore (Nos. RG 107/19, RG 11/20 and RT 14/20), and the Agency for Science, Technology and Research (A*STAR) under its MTC Individual Research Grants (No. M21K2c0114).
J.D. Winkler, M.B. Rouse, M.F. Greaney, et al., J. Am. Chem. Soc. 124 (2002) 9726–9728. doi: 10.1021/ja026600a
E.E. van Tamelen, T.A. Spencer, D.S. Allen, R.L. Orvis, J. Am. Chem. Soc. 81 (1959) 6341–6342. doi: 10.1021/ja01532a070
Q. Xiao, W.W. Ren, Z.X. Chen, et al., Angew. Chem. Int. Ed. 50 (2011) 7373–7377. doi: 10.1002/anie.201103088
T. Shi, X. Li, Y. Li, et al., Green Synth. Catal. 4 (2023) 58–63. doi: 10.1016/j.gresc.2022.05.008
C. Hui, A.P. Antonchick, Green Synth. Catal. 3 (2022) 339–348. doi: 10.1016/j.gresc.2022.07.001
T.P. Majhi, B. Achari, P. Chattopadhyay, Heterocycles 71 (2007) 1011–1052. doi: 10.3987/REV-07-612
F. Kopp, C.F. Stratton, L.B. Akella, D.S. Tan, Nat. Chem. Biol. 8 (2012) 358–365. doi: 10.1038/nchembio.911
R.A. Bauer, T.A. Wenderski, D.S. Tan, Nat. Chem. Biol. 9 (2013) 21–29. doi: 10.1038/nchembio.1130
T.V. Nguyen, J.M. Hartmann, D. Enders, Synthesis 45 (2013) 845–873. doi: 10.1055/s-0032-1318152
A.H. Shoemaker, D.R. Griffith, Synthesis 53 (2021) 65–78. doi: 10.1055/s-0040-1707385
J. H Fan, Y.J. Hu, L.X. Li, et al., Nat. Prod. Rep. 38 (2021) 1821–1851. doi: 10.1039/D1NP00003A
I.U. Khand, G.R. Knox, P.L. Pauson, W.E. Watts, J. Chem. Soc., Perkin Trans. 1 (1973) 975–977.
I.U. Khand, G.R. Knox, P.L. Pauson, et al., J. Chem. Soc., Perkin Trans. 1 (1973) 977–981.
Z. Yang, Acc. Chem. Res. 54 (2021) 556–568. doi: 10.1021/acs.accounts.0c00709
M.M. Heravi, L. Mohammadi, RSC Adv. 11 (2021) 38325-38373. doi: 10.1039/D1RA05673E
S. Chen, C. Jiang, N. Zheng, Z. Yang, L. Shi, Catalysts 10 (2020) 1199. doi: 10.3390/catal10101199
Z. Zhang, D. Zhao, Y. He, et al., Chin. Chem. Lett. 30 (2019) 1503–1505. doi: 10.1016/j.cclet.2019.03.033
C.C. Liu, C. Lei, Y. Zhong, et al., Tetrahedron Lett. 70 (2014) 4317–4322. doi: 10.1016/j.tet.2014.05.019
T. Ma, H. Cheng, M. Pitchakuntla, W. Ma, Y. Jia, J. Am. Chem. Soc. 144 (2022) 20196–20200. doi: 10.1021/jacs.2c08694
H.Y. Tian, L.J. Ruan, T. Yu, et al., J. Nat. Prod. 80 (2017) 1182–1186. doi: 10.1021/acs.jnatprod.6b01018
M.J. Cheng, L.P. Zhong, C.C. Gu, et al., J. Am. Chem. Soc. 142 (2020) 12602–12607. doi: 10.1021/jacs.0c05479
Y. Wang, B. Chen, X. He, J. Gui, J. Am. Chem. Soc. 142 (2020) 5007–5012. doi: 10.1021/jacs.0c00363
Y. Wang, H. Tian, J. Gui, J. Am. Chem. Soc. 143 (2021) 19576–19586. doi: 10.1021/jacs.1c10067
Y. Wang, W. Ju, H. Tian, W. Tian, J. Gui, J. Am. Chem. Soc. 140 (2018) 9413–9416. doi: 10.1021/jacs.8b06444
Y. Wang, W. Ju, H. Tian, et al., J. Am. Chem. Soc. 141 (2019) 5021–5033. doi: 10.1021/jacs.9b00925
J. Huang, T. Cao, Z. Zhang, Z. Yang, J. Am. Chem. Soc. 144 (2022) 2479–2483. doi: 10.1021/jacs.1c12395
J. Gao, P. Rao, K. Xu, et al., J. Am. Chem. Soc. 142 (2020) 4592–4597. doi: 10.1021/jacs.0c00308
C. He, J. Xuan, P. Rao, et al., Angew. Chem. Int. Ed. 58 (2019) 5100–5104. doi: 10.1002/anie.201900782
H. Chu, J.M. Smith, J. Felding, P.S. Baran, ACS Cent. Sci. 3 (2017) 47–51. doi: 10.1021/acscentsci.6b00313
B. Wang, B. Xu, W. Xun, et al., Angew. Chem. Int. Ed. 60 (2021) 9439–9443. doi: 10.1002/anie.202016212
B.M. Gross, S.J. Han, S.C. Virgil, B.M. Stoltz, J. Am. Chem. Soc. 145 (2023) 7763–7767. doi: 10.1021/jacs.3c02142
W.L. Xiao, R.T. Li, S.X. Huang, J.X. Pu, H.D. Sun, Nat. Prod. Rep. 25 (2008) 871–891. doi: 10.1039/b719905h
J. Zhou, G. Zhan, H. Zhang, et al., Org. Lett. 19 (2017) 3935–3938. doi: 10.1021/acs.orglett.7b01863
U. Rasmussen, S.B. Christensen, F. Sandberg, Acta Pharm. Suec. 15 (1978) 133–140.
U.W. Smitt, S.B. Christensen, Planta Med. 57 (1991) 196–197. doi: 10.1055/s-2006-960067
H. Liu, K.G. Jensen, L.M. Tran, et al., Phytochemistry 67 (2006) 2651–2658. doi: 10.1016/j.phytochem.2006.10.005
A.G. Kozlovsky, V.P. Zhelifonova, S.M. Ozerskaya, et al., Pharmazie 55 (2000) 470–471.
H. Yan, G.S. Smith, F.E. Chen, Green Synth. Catal. 3 (2022) 219–226. doi: 10.1016/j.gresc.2022.05.007
H. Zhang, S.L. Shyaula, J.Y. Li, J. Li, J.M. Yue, Org. Lett. 18 (2016) 1202–1205. doi: 10.1021/acs.orglett.6b00362
S. Shen, H. Zhu, D. Chen, et al., Tetrahedron Lett. 53 (2012) 5759–5762. doi: 10.1016/j.tetlet.2012.08.049
C.Y. Duh, S.K. Wang, M.C. Chia, M.Y. Chiang, Tetrahedron Lett. 40 (1999) 6033–6035. doi: 10.1016/S0040-4039(99)01194-6
J.P. Tuccinardi, J.L. Wood, J. Am. Chem. Soc. 144 (2022) 20539–20547. doi: 10.1021/jacs.2c09826
C.P. Jasperse, D.P. Curran, T.L. Fevig, Chem. Rev. 91 (1991) 1237–1286. doi: 10.1021/cr00006a006
M. Malacria, Chem. Rev. 96 (1996) 289–306. doi: 10.1021/cr9500186
M. Yan, J.C. Lo, J.T. Edwards, P.S. Baran, J. Am. Chem. Soc. 138 (2016) 12692–12714. doi: 10.1021/jacs.6b08856
Y. Zhang, K. Sun, Q. Lv, et al., Chin. Chem. Lett. 30 (2019) 1361–1368. doi: 10.1016/j.cclet.2019.03.034
A. Turlik, Y. Chen, A.C. Scruse, T.R. Newhouse, J. Am. Chem. Soc. 141 (2019) 8088–8092. doi: 10.1021/jacs.9b03751
M. Hergenhahn, W. Adolf, E. Hecker, Tetrahedron Lett. 16 (1975) 1595–1598.
A. Szallasi, P.M. Blumberg, Neuroscience 30 (1989) 515–520. doi: 10.1016/0306-4522(89)90269-8
W. Adolf, B. Sorg, M. Hergenhahn, E. Hecker, J. Nat. Prod. 45 (1982) 347–354. doi: 10.1021/np50021a018
S. Hashimoto, S. Katoh, T. Kato, D. Urabe, M. Inoue, J. Am. Chem. Soc. 139 (2017) 16420–16429. doi: 10.1021/jacs.7b10177
B.M. Trost, C. Min, Nat. Chem. 12 (2020) 568–573. doi: 10.1038/s41557-020-0439-y
D. Chen, P.A. Evans, J. Am. Chem. Soc. 139 (2017) 6046–6049. doi: 10.1021/jacs.7b01734
R.F. Lusi, G. Sennari, R. Sarpong, Nat. Chem. 14 (2022) 450–456. doi: 10.1038/s41557-021-00870-4
S.E. Denmark, J. Fu, Chem. Rev. 103 (2003) 2763–2794. doi: 10.1021/cr020050h
T.P. Loh, J.R. Zhou, Z. Yin, Org. Lett. 1 (1999) 1855–1857. doi: 10.1021/ol990316f
X. Hu, S. Xu, T.J. Maimone, Angew. Chem. Int. Ed. 56 (2017) 1624–1628. doi: 10.1002/anie.201611078
N.O. Calloway, Chem. Rev. 17 (1935) 327–392. doi: 10.1021/cr60058a002
X. Wei, I.I. Rodríguez, A.D. Rodríguez, C.L. Barnes, J. Org. Chem. 72 (2007) 7386–7389. doi: 10.1021/jo070649n
H.D. Hao, D. Trauner, J. Am. Chem. Soc. 139 (2017) 4117–4122. doi: 10.1021/jacs.7b00234
L. Yet, Chem. Rev. 100 (2000) 2963–3008. doi: 10.1021/cr990407q
A. Michaut, J. Rodriguez, Angew. Chem. Int. Ed. 45 (2006) 5740–5750. doi: 10.1002/anie.200600787
M.E. Maier, Angew. Chem. Int. Ed. 39 (2000) 2073–2077. doi: 10.1002/1521-3773(20000616)39:12<2073::AID-ANIE2073>3.0.CO;2-0
A. Deiters, S.F. Martin, Chem. Rev. 104 (2004) 2199–2238. doi: 10.1021/cr0200872
M.D. McReynolds, J.M. Dougherty, P.R. Hanson, Chem. Rev. 104 (2004) 2239–2258. doi: 10.1021/cr020109k
P.P. Zhang, Z.M. Yan, Y.H. Li, J.X. Gong, Z. Yang, J. Am. Chem. Soc. 139 (2017) 13989–13992. doi: 10.1021/jacs.7b07388
J.L. von Salm, C.G. Witowski, R.M. Fleeman, et al., Org. Lett. 18 (2016) 2596–2599. doi: 10.1021/acs.orglett.6b00979
T. Siemon, S. Steinhauer, M. Christmann, Angew. Chem. Int. Ed. 58 (2019) 1120–1122. doi: 10.1002/anie.201813142
L. Min, Y.J. Hu, J.H. Fan, et al., Chem. Soc. Rev. 49 (2020) 7015–7043. doi: 10.1039/D0CS00365D
Y.J. Hu, L.X. Li, J.C. Han, et al., Chem. Rev. 120 (2020) 5910–5953. doi: 10.1021/acs.chemrev.0c00045
L. Min, X. Liu, C.C. Li, Acc. Chem. Res. 53 (2020) 703–718. doi: 10.1021/acs.accounts.9b00640
T. Amagata, A. Amagata, K. Tenney, et al., Org. Lett. 5 (2003) 4393–4396. doi: 10.1021/ol0356800
J. Liu, J. Wu, J.H. Fan, et al., J. Am. Chem. Soc. 140 (2018) 5365–5369. doi: 10.1021/jacs.8b02629
S. Yamamura, J.A. Lamberton, M. Niwa, K. Endo, Y. Hirata, Chem. Lett. 9 (1980) 393–396. doi: 10.1246/cl.1980.393
L.X. Li, L. Min, T.B. Yao, et al., J. Am. Chem. Soc. 144 (2022) 18823–18828. doi: 10.1021/jacs.2c09548
H.Y. Tian, L. Wang, X.Q. Zhang, et al., Chem. Eur. J. 16 (2010) 10989–10993. doi: 10.1002/chem.201000847
L.P. Zhong, R. Feng, J.J. Wang, C.C. Li, J. Am. Chem. Soc. 145 (2023) 2098–2103. doi: 10.1021/jacs.2c13494
D.D. Zhang, J.B. Xu, Y.Y. Fan, et al., J. Org. Chem. 85 (2020) 3742–3747. doi: 10.1021/acs.joc.9b03310
Y. P Zou, Z.L. Lai, M.W. Zhang, et al., J. Am. Chem. Soc. 145 (2023) 10998–11004. doi: 10.1021/jacs.3c03755
S. Felder, S. Dreisigacker, S. Kehraus, et al., Chem. Eur. J. 19 (2013) 9319–9324. doi: 10.1002/chem.201301379
M. Schmid, A. Grossmann, K. Wurst, T. Magauer, J. Am. Chem. Soc. 140 (2018) 8444–8447. doi: 10.1021/jacs.8b06228
J. Sheu, F. Ahmed, R. Shiue, C. Dai, Y. Kuo, J. Nat. Prod. 65 (2002) 1904–1908. doi: 10.1021/np020280r
N.J. Hafeman, S.A. Loskot, C.E. Reimann, et al., J. Am. Chem. Soc. 142 (2020) 8585–8590. doi: 10.1021/jacs.0c02513
Y. Li, Y.B. Liu, Y.L. Liu, et al., Org. Lett. 16 (2014) 4320–4323. doi: 10.1021/ol5020653
Y. Wang, R. Zhao, M. Yang, J. Am. Chem. Soc. 144 (2022) 15033–15037. doi: 10.1021/jacs.2c06981
L. Kong, H. Yu, M. Deng, et al., J. Am. Chem. Soc. 144 (2022) 5268–5273. doi: 10.1021/jacs.2c01692

Scheme 4 Miscellaneous methods for total synthesis of bufospirostenin A by Gui and Yang (2021 and 2022).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

























