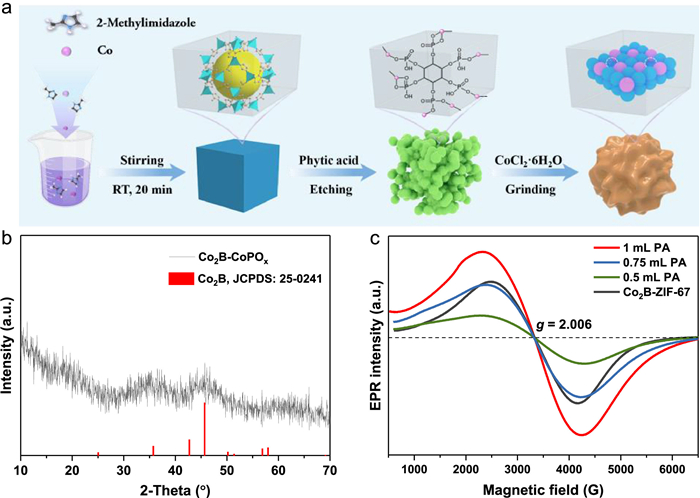
Figure 1.
(a) Schematic diagram for the preparation of Co2B-CoPOx. (b) XRD pattern of Co2B-CoPOx. (c) EPR spectra of as-synthesized samples at different volume of PA.
Phytic acid-derivative Co2B-CoPOx coralloidal structure with delicate boron vacancy for enhanced hydrogen generation from sodium borohydride
Luyan Shi , Ke Zhu , Yuting Yang , Qinrui Liang , Qimin Peng , Shuqing Zhou , Tayirjan Taylor Isimjan , Xiulin Yang

Hydrogen-related energy devices, including hydrogen-oxygen fuel cells, large-scale hydrogen power generation, and other hydrogen-powered devices, have incentivized the revolution of various hydrogen evolution technologies [1]. In particular, hydrogen storage materials catalytic hydrogen (H2) generation technology has been deemed an up-and-coming trend to realize a sustainable energy scheme [2]. Among the chemical storage sources of hydrogen, metal hydrides such as NaBH4, LiAlH4 and MgH2 have garnered tremendous recent attention [3–5]. Among them, NaBH4 is emerging as a central focus with a theoretical gravimetric hydrogen storage capacity of 10.6 wt%, excellent hydrolysis controllability, milder operation conditions, and high hydrogen purity, which can be applied to hydrogen production by hydrolysis [6,7]. Moreover, NaBH4 can facilely regenerate from its hydrolytic product, making it possible for the NaBH4-H2-PEMFC system to be a primary source device for on-demand power supply [8,9]. These advantages yield NaBH4 hydrolysis hydrogen production a bright future in real-world applications. Nevertheless, several hurdles need to be addressed before its commercial deployment, including the sluggish kinetic of NaBH4 self-hydrolysis and low H2 yield [10]. Exploring efficient catalysts is a feasible strategy for overcoming kinetic barriers for the hydrolysis of NaBH4. The ideal hydrolysis reaction of NaBH4 in the presence of a catalyst is given as Eq. 1 [11]:
|
|
(1) |
However, considerable market barriers still exist because the noble metals (e.g., Ru, Pt, Pd) are the most widely applied catalysts to negotiate the hydrolysis of NaBH4, resulting in poor cost competitiveness [12,13]. Alternatively, recent progress in the search of new catalysts has proven that a range of Co-based borides and phosphides are attractive alternatives. Especially, the active sites of Co-based borides have been well identified, which can stimulate the strategy to tailor Co-based borides for promoted NaBH4 hydrolysis by modulating material parameters such as vacancies and the electronic interaction between transition metal atoms and boron atoms [14,15]. In addition, high electronegative heteroatom doping can increase the number and modulate the electronic surrounding of metal active sites, thus improving the activities of the catalyst [16]. For example, Mehdi et al. reported P-induced Co-based monolithic efficient catalysts for NH3BH3 hydrolysis and showed that the induction of P caused electron transfer from Co to P, promoting new active sites for the efficient adsorption of reactant molecules [17]. Notably, the highly electronegative phosphorus (P) atom possesses lone-pair electrons in 3p orbitals and vacant 3d orbitals, which can mediate local charge density and change the surface charge state, further advancing the catalytic performance [18]. We assume that the P-induced approach also has a positive effect on the catalyst of NaBH4 hydrolysis. However, conventional procedures (gas-solid reaction scheme) use NaH2PO2 as the P source, which would release poisonous and auto-ignition PH3 products into the environment [19]. Phytic acid (PA, myo-inositol 1,2,3,4,5,6-hexakisphosphate) is a naturally environmentally friendly and renewable biological compound, and its six phosphate groups can easily chelate to different metal ions, which can serve as an ideal P source to replace NaH2PO2 [20]. In the processes of PA etching, P possesses higher electronegativity than metallic (M) atoms, which tend to receive electrons from the metal, causing a redistribution of the electronic properties of the catalysts and further forming the key active sites favoring the interaction and adsorption with reaction intermediates [21]. On the other hand, vacancy engineering is considered a new "intrinsic" strategy to enhance catalytic activity by inducing high distortion energy and diverse atomic rearrangements [22]. Besides, vacancies can manipulate the electronic band structure as well as lower the activation energy of adsorption and dissociation of reactants [23]. Consequently, the electronic structure and surface nature of the catalyst are two versatile knobs for enhancing the intrinsic catalytic activity, which can be favorably tailored through doping and vacancy engineering.
In this work, a light P doping and rich boron vacancies Co2B-CoPOx was developed as a highly efficient catalyst for NaBH4 hydrolysis. The coralloidal structure of Co2B-CoPOx is porous with an average pore size of about 23.8 nm. Such unique structural features not only provide more active sites but also may improve electron transfer dynamics, beneficial to the hydrolysis of NaBH4. Moreover, the experimental and characteristic analyses show that the doping of P tunes the relative content of B and induces more boron vacancies, which subsequently affects the electronic structure and facilitates the reversible dissociation of B-H on active sites. As a result, Co2B-CoPOx exhibits excellent hydrolysis activity and scalable reusability. Importantly, the prepared robust Co2B-CoPOx catalyst reveals remarkably efficient hydrogen evolution from NaBH4 in comparison to previous non-noble catalysts and even noble catalysts presented in the literature. Our work provides new opportunities for designing advanced metal borides catalysts for the hydrolysis of NaBH4 toward improved viability in generating H2 from NaBH4.
The synthesis procedure of Co2B-CoPOx is elaborated in Fig. 1a. Initially, uniform Co-based zeolitic imidazolate framework-67 (ZIF-67) nanocubes (NCs) were synthesized using a surfactant-assisted approach. The successful synthesis of ZIF-67 NCs was confirmed by the X-ray diffraction (XRD) pattern in Fig. S1 (Supporting information), which matched well with the simulated ZIF-67 [24]. Forming the chelation structure of P-O-Co in the precursor is notoriously challenging that requires pertinent chelation structure engineering. We present a PA-assisted method to achieve such goal, where PA coordinated with Co2+ through its six negatively charged phosphate groups to form Co-PA. During this process, PA acted as an organic acid to release proton, breaking the chelation of Co2+ and 2-methylimidazole, resulting in the loss of a portion of the organic ligands in ZIF-67. Simultaneously, PA also coordinated with Co2+ through P-O-M and served as a P source. The chemical etching and coordination reaction in the PA solution transformed ZIF-67 into amorphous Co-PA, as observed from the disappearance of ZIF-67 peaks and the formation of amorphous Co-PA in Fig. S1. Next, Co-PA was ground with CoCl2·6H2O and urea, followed by boronation using NaBH4, to convert it into Co2B-CoPOx. The XRD pattern of Co2B-CoPOx in Fig. 1b confirmed that all the diffraction peaks can be indexed well to Co2B (JCPDS No. 25-0241). The weak and broad diffraction peaks indicated that the Co2B crystallites were in the nanometer size range, and the crystallinity was weak [25]. Control samples were also characterized using XRD. Fig. S2a (Supporting information) showed that the diffraction pattern of Co2B-ZIF-67 was similar to that of ZIF-67 but slightly protruded at 30°−40° and 40°−50°, indicating the presence of Co2B in the sample. Fig. S2b (Supporting information) confirmed the formation of the control sample Co2B (JCPDS No. 25-0241). Figs. S3a and b (Supporting information) showed that ZIF-67-urea and Co-PA-urea were the amorphous structure.
Fourier transform infrared (FT-IR) measurement was conducted to probe the local binding structure of Co during the reaction process (Fig. S4 in Supporting information). The peaks observed around 423 cm−1 correspond to the Co-N bonding of 2-methylimidazole, while the peaks around 2900 cm−1 are associated with the C—H stretching vibration of the methyl group [26]. After the chemical etching in PA solution, the intensity of these peaks diminished in both Co-PA and Co2B-CoPOx, revealing the substitution of 2-methylimidazole by PA and the formation of PA cross-linked Co complexes [27]. The N2 absorption/desorption isotherms of Co2B-CoPOx and Co2B-ZIF-67 are shown in Fig. S5 (Supporting information), exhibiting a typical type Ⅳ isotherm, suggesting the presence of mesopores. The Brunauer-Emmett-Teller (BET) surface area of Co2B-CoPOx was calculated to be 15.5 m2/g, which is lower than that of Co2B-ZIF-67 (736.3 m2/g). The decreased BET specific surface area can be ascribed to the partially disrupted structure of ZIF-67 in phytic acid solution. The reduction in surface area further supports the coordination of Co2+ with PA, which predicts improved electronic structure [28,29]. Additionally, the pore size distribution of Co2B-CoPOx is 23.8 nm, significantly larger than that of Co2B-ZIF-67 (2.6 nm), which well endows Co2B-CoPOx with higher catalytic activity by providing more active sites and helping the H2 to diffuse along the surface (the inset of Fig. S5) [30]. Electron paramagnetic resonance (EPR) has been proved to be an effective tool to characterize the vacancies in the catalysts. As depicted in Fig. 1c, a distinct signal around g = 2.006 was observed, which can be assigned to the unpaired electrons trapped by boron vacancies [31]. A monotonic increase in the boron vacancy concentration is observed with increasing volume of PA. It should be noted that the B content decreased from 11.9 wt% for Co2B-ZIF-67 to 9.2 wt% for Co2B-CoPOx, as confirmed by inductively coupled plasma mass spectroscopy (ICP-MS) measurements (Table S1 in Supporting information). This suggests that phosphorization after PA treatment resulted in a slight loss of B atoms and the creation of more boron vacancies [32,33]. Additionally, the introduction of P species into ZIF-67 lead to a different bonding environment for Co-O-P bonds compared to original N—Co-N structure, which contributed to the disorder of lattice cell and the formation of more boron vacancies [34]. The increased number of boron vacancies in Co2B-CoPOx substantially modify its electronic structure and provided active sites for NaBH4 adsorption, leading to enhanced catalytic activity [35].
The morphological evolution of Co2B-CoPOx composite was tracked using scanning electron microscope (SEM). Fig. S6a (Supporting information) shows that the initially formed purple ZIF-67 consists exclusively of uniform nanocubes. After PA etching, the SEM image reveals that the obtained pink Co-PA exhibits a nanoparticles morphology with rough edges, and the nanoparticles are in contact with each other (Fig. S6b in Supporting information). These significant morphological changes are related to the dissolution of organic ligands, which causes the disintegration and reorganization of the nanocubes structure. As the boronizing process proceeds, a well-developed, defined coral-like three-dimensional (3D) morphology of Co2B-CoPOx is formed (Fig. 2a). This coral-like infrastructure favors the close contact of reactants with active sites and promotes H2 diffusion [36]. In contrast, Co2B-ZIF-67 still inherits the cubic morphology (Fig. S7 in Supporting information), indicating the crucial role of PA in the formation of the coral-like structure of Co2B-CoPOx. The microstructure of Co2B-CoPOx is further examined by transmission electron microscopy (TEM). The low-magnification TEM image shows that Co2B-CoPOx presents a porous structure (Fig. 2b). Moreover, the high-resolution TEM (HR-TEM) image reveals the absence of well-resolved lattice fringes (Fig. 2c). The corresponding selected area electron diffraction (SADE) pattern exhibits halo rings (Fig. 2c, inset), verifying the formation of a weak crystalline structure that is consistent with the XRD results. The energy dispersive X-ray (EDX) pattern proves the co-existence of Co, B, P, and O elements (Fig. S6c in Supporting information). The high-angle annular dark-field scanning TEM (HAADF-STEM) and EDX mapping images demonstrate the homogeneous distribution of Co, B, P, and O elements (Fig. S6d in Supporting information).
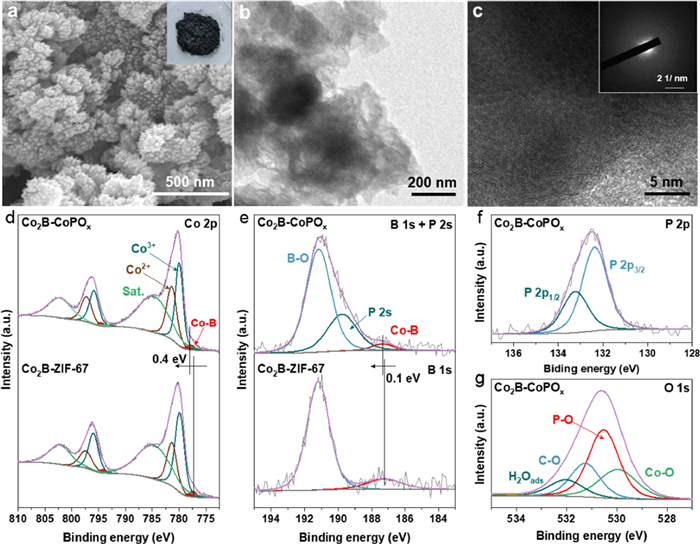
The practical structure and elemental chemical state of Co2B-CoPOx were elucidated by X-ray photoelectron spectroscopy (XPS). The XPS survey spectrum of Co2B-CoPOx corroborated the presence of Co, B, P and O elements (Fig. S8a in Supporting information), while only Co, B and O were observed in Co2B-ZIF-67, manifesting the successful doping of P. The high-resolution C 1s spectra were deconvoluted into four peaks corresponding to C=C (284.0 eV), C−C (284.8 eV), C−O (286.0 eV) and C=O (288.5 eV) (Figs. S8b and c in Supporting information), which served as a calibration standard [37]. The Co 2p XPS spectrum exhibited two blunt satellite peaks and two sharp peaks, corresponding to the spin-orbit splitting of the Co 2p3/2 and 2p1/2 components, respectively (Fig. 2d) [38]. The two prominent peaks at 779.9 eV and 781.3 eV were assigned to Co3+ and Co2+ in the Co 2p3/2 region [39,40]. Simultaneously, the other two fitted peaks at around 777.8 and 784.7 eV were attributed to Co-B bonding and shake-up satellites, respectively [41]. The coexistence of Co3+ and Co2+ suggests a fraction of Co3+ is reduced to Co2+ by excessive NaBH4 to form B vacancies [42]. In Fig. 2e, the characteristic peaks at 187.2 and 191.2 eV corresponded to Co-B and B-O bonding in Co2B-ZIF-67 [43]. In Co2B-CoPOx, a slight positive shift of Co-B bond was observed, and the appearance of the P 2s peak indicated the presence of phosphate. The positive shift demonstrates a drastic electronic coupling between Co-B and phosphate, where phosphate has strong electronegativity and can draw electrons from Co-B species [44,45]. The P 2p high-resolution XPS spectrum of Co2B-CoPOx was disassembled into two sets of peaks corresponding to P 2p3/2 and P 2p1/2 (Fig. 2f), further validating the successful induction of phosphate in the Co2B-CoPOx catalyst [46]. The O 1s spectra of Co2B-CoPOx (Fig. 2g) showed peaks at approximately 529.9, 530.5, 531.3, and 532.0 eV, which can be considered as Co-O, P-O, C—O and H2Oads, respectively [47,48]. As depicted in Fig. S9 (Supporting information) Co2B-CoPOx (19.3) has a smaller contact angle compared to Co2B-ZIF-67 (51.2), elucidating that Co2B-CoPOx possesses higher hydrophilicity. The presence of a mass of hydroxyl groups enhances the contact between the NaBH4 solution and the catalyst, thereby improving the catalytic performance [49]. Overall, the introduction of phosphate species and B vacancies effectively regulates the electronic structure and local coordination environment of Co centers, thereby accelerating electron transport and boosting the hydrolysis of NaBH4 [50]. Consequently, Co2B-CoPOx possesses superior catalytic activity compared to Co2B-ZIF-67, with catalysis results discussed below later [51].
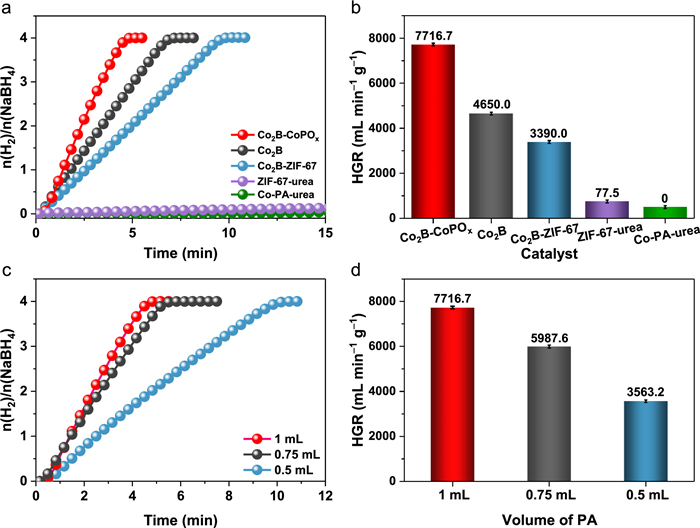
The catalytic activities were assessed in an alkaline NaBH4 solution at 25 ℃ using a water displacement method, as illustrated in Fig. S10 (Supporting information). To investigate the role of P doping in improving NaBH4 hydrolysis for H2 production, we carried out control experiments using weak acid without P (oxalic acid) for etching the catalysts and investigated the difference in performance. As shown in Figs. S11a and b (Supporting information), catalysts etched with the same pH of oxalic acid exhibited poor hydrolysis activity, with an HGR value of 1881.5 mL min−1 g−1. This is in sharp contrast to the Co2B-CoPOx etched with PA, which displayed significant catalytic activity with an HGR value of 7716.7 mL min−1 g−1. This fully confirms that P is a crucial factor in determining the catalyst's high activity. In addition, somewhat delayed hydrogen evolution was observed for all samples, due to initial catalyst wetting and pore diffusion resistance [52].
As exhibited in Figs. 3a and b, directly grinding ZIF-67 and Co-PA resulted in impaired activity, suggesting that effective active sites of Co2B cannot be formed through this method. However, after PA treatment and the second step of boronation, Co2B-CoPOx exhibited remarkably enhanced catalytic activity, completing the hydrolysis of NaBH4 in only 4.8 min with an HGR value of 7716.7 mL min−1 g−1. In comparison, Co2B-ZIF-67 and Co2B showed inferior activity, requiring 9.7 min (HGR = 3390.0 mL min−1 g−1) and 7.0 min (HGR = 3390.0 mL min−1 g−1), respectively, to complete the full reaction. This highlights the critical impact of P doping and B vacancy engineering on the catalytic property of the material. To optimize the proportion of PA, different volumes (0.5, 0.75, 1, 1.25 and 1.5 mL) of PA were introduced into the ethanol solution. Figs. 3c and d show that 1 mL of PA possessed the most remarkable hydrogen generation rate, while 1.25 mL and 1.5 mL of PA exhibited inactive behavior. The above phenomenon can be attributed to the following reasons: when the volume of PA is 0.5 and 0.75 mL, the solution appears purple and light pink-purple, respectively, indicating insufficient etching of ZIF-67 by PA and leading to a small HGR value (Fig. S12 in Supporting information). On the other hand, as the volume of PA increases from 1.25 to 1.5 mL, the solution turns brown or even transparent, demonstrating the complete dissolution of ZIF-67 due to the high concentration of H+ induced by a large amount of PA. The surface vacancies of catalysts may induce positive effects on the hydrogen generation of NaBH4. We next tried to quantify the boron vacancies by XPS to unveil the vacancy-activity relationship. The content ratio was calculated based on the proportion of its fitting peaks [53]. The EPR spectra demonstrated the concentration of boron vacancies can be modulated by phosphorus doping content [54]. The content ratio of P is 29.0%, 21.1% and 17.2% for Co2B-CoPOx-1 mL, Co2B-CoPOx-0.75 mL and Co2B-CoPOx-0.5 mL, respectively (Fig. S13 in Supporting information). It can be seen the general increase in boron vacancies concentration with the increase of PA. Then, we investigated the effect of boron content on hydrolysis of NaBH4. Fig. 3d showed that the HGR value was apparent increase when the volume of PA was increased from 0.5 mL to 1 mL. Hence, more boron vacancies can contribute the hydrolysis of NaBH4.
NaOH is usually introduced as a stabilizer into the NaBH4 hydrolysis system to prevent self-hydrolysis. In the absence of a catalyst, self-hydrolysis of 150 mmol/L NaBH4 solution takes about 10 h to produce 110 mL of H2 and negligible hydrogen gas is produced from 150 mmol/L NaBH4 + 0.4 wt% NaOH solution, verifying the stability of NaBH4 in water and an alkaline environment (Fig. S14 in Supporting information). To further explore the effect of NaOH, hydrogen production with different concentrations of NaOH was carried out under the same conditions. Fig. S15a (Supporting information) shows that the addition of NaOH significantly promotes the catalytic hydrolysis of NaBH4, and the H2 generation rate barely changed as NaOH concentrations increased from 0.4 wt% to 3 wt%. However, a further increase of NaOH concentration (from 3 wt% to 10 wt%) leads to a decrease in HGR. As reported by others, the hydrolysis mechanism of NaBH4 differs on utilization of different catalysts [55,56]. In this work, the OH− ions play a dual role during the catalytic hydrolysis reaction. Appropriate concentration of OH− ions can activate active sites, thereby enhancing catalytic activity. The higher NaOH concentration results in the increase in viscosity and the alkalinity of the solution, inhibiting the hydrolysis reaction [57]. Therefore, 0.4 wt% NaOH was selected as the optimal concentration to avoid reagent waste. The influence of NaBH4 concentrations on the hydrolysis catalyzed by Co2B-CoPOx was also investigated (Fig. S15b in Supporting information). The H2 generation rate versus the concentration of NaBH4 is plotted on a natural logarithmic scale in the inset of Fig. S15b. The slop of the logarithmic plot is 0.083, indicating a zero-order reaction with respect to NaBH4 concentration [58]. This unveils that NaBH4 is easily activated, and its activation is not the rate-determining step in the hydrolysis process [59]. The amount of catalyst is another decisive factor in determining the catalytic reaction rate. As seen in Figs. S15c and d (Supporting information), the hydrogen generation rate of Co2B-CoPOx varied with the change in catalyst mass from 5 mg to 20 mg. Among the series, 10 mg of Co2B-CoPOx exhibited the highest hydrogen generation rate, making it the optimal amount of catalyst and resulting in substantial cost reduction.
The effect of P doping on the activation energy in NaBH4 hydrolysis was explored by investigating the temperature dependence of hydrogen generation kinetics at different temperatures, as shown in Fig. 4a and Fig. S16a (Supporting information). Based on the Arrhenius plots, the activation energy (Ea) value of Co2B-CoPOx was calculated to be 44.9 kJ/mol, which is lower than that of Co2B-ZIF-67 (56.1 kJ/mol) (Fig. S16b in Supporting information), and significantly lower than the values reported for most non-noble or noble mental catalysts in previous literature (Fig. 4b). This validates that P doping effectively reduced the energy barrier for molecule activation. Hydrogen generation with NH3BH3 is proceeded under a similar method. A HGR of 1891.9 mL min−1 g−1 is achieved (Fig. S17 in Supporting information). Compared with NaBH4 (7716.7 mL min−1 g−1), a decreased activity is emerged for NH3BH3 hydrolysis. This negative activity is attributed to the discrepant catalytic mechanism during borohydride hydrolysis caused by the different molecular geometry of NaBH4 and NH3BH3 [60]. The reusability is very important for catalysts in the aspect of the practical application. Therefore, the reusability of Co2B-CoPOx catalyst was evaluated through a continuous recycling test of catalytic NaBH4 hydrolysis. As depicted in Figs. 4c and d, the catalyst exhibited a slight degradation in activity after 5 cycles. As demonstrated in Fig. S18 (Supporting information), the XRD pattern shows that the Co2B-CoPOx after 5 cycles keeps a similar structural composition. Moreover, XPS analysis (Fig. S19 in Supporting information) exhibits that the original elemental composition, chemical state and the peak intensities were similar to Co2B-CoPOx even after 5 cycles. These results clearly indicate that our developed catalyst bears a stable coralloidal structure. The SEM images show that the catalyst's surface occurs agglomerations after catalyzing the hydrolysis of NaBH4 (Fig. S20 in Supporting information). Therefore, the slight decrease in catalytic activity can be attributed to surface agglomeration and accumulation of metaborate on the catalyst surface, resulting in a reduction of boron vacancies and thus hindering electron transmission [61,62]. Overall, the Co2B-CoPOx catalyst demonstrated outstanding activity and reusability, making it cost-effective and suitable for practical industrial applications.
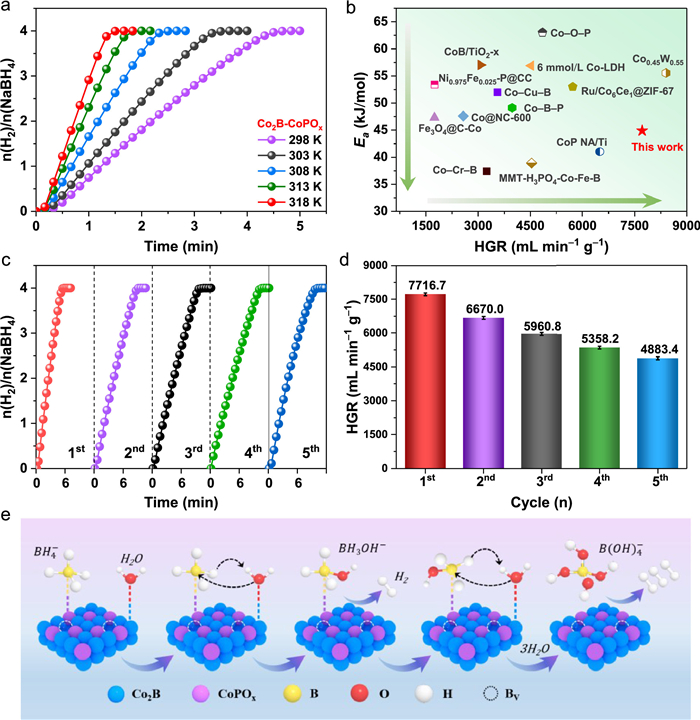
We next tried to shed light on the underlying origin of the striking catalysis performance of Co2B-CoPOx based on Langmuir-Hinshelwood model and many previous literatures, the plausible mechanism for NaBH4 hydrolysis over Co2B-CoPOx catalyst is shown in Eqs. 2–5 and Fig. 4e [55,61,63]. According to XPS study, P species, being strongly electronegative, draw electrons from Co2B, resulting in a higher electron density on CoPOx species compared to Co2B species. Consequently, the B atoms with a few positive charges in BH4− ions are adsorbed by the electron-rich CoPOx species through the binding sites provided by boron vacancies, while the electron-deficient Co2B species adsorb H2O molecules. In step 1, the adsorbed water molecule attacks BH4−. Consequently, one molecule of H2 is released and an adsorbed BH3OH− is lefts on the surface. In step 2, another adsorbed H2O molecule attacks BH3OH−, and the second H2 molecule is released and the BH2(OH)2− is produced and lefts on the CoPOx species surface. In step 3, a new H2O molecule adsorbs at Co2B species and attacks BH2(OH)2− to produce BH(OH)3− and H2. In step 4, H2O molecule attacks BH(OH)3− to give H2 and B(OH)4−. Meanwhile, the B(OH)4− is desorbs. Therefore, the hydrolysis reaction ends with compete conversion of BH4− ion to B(OH)4− ion with release of four H2 molecules [64]. This mechanism highlights the role of P doping and B vacancy engineering in the Co2B-CoPOx catalyst, where P enhances the electron density of CoPOx species, facilitating the adsorption of B atoms through boron vacancies. This unique interaction between the catalyst and reactants promotes the efficient hydrolysis of NaBH4, leading to enhanced catalytic activity and hydrogen generation.
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
In conclusion, we report that the P doping rich boron vacancy on Co2B-CoPOx can be achieved through chelating Co with negatively charged phosphate groups. The rational P doping strategy simultaneously realized the high-level chemical induction and defects in the catalyst. The presence of abundant boron vacancy enhances the catalyst's capacity to capture and interact with reaction intermediates effectively. The high electronegativity of P species serves as an electronic structure modifier to tune the local charge of Co via electron transfer, creating favorable condition for the dissociation of B-H and O—H bonds and rendering impressive catalytic activity. The hydrolysis of NaBH4 measurements certify the exceptional activity of Co2B-CoPOx, with a hydrogen generation rate (HGR) of 7716.7 mL min−1 g−1, which is 2.3 times higher than that of Co2B-ZIF-67. Furthermore, Co2B-CoPOx exhibited superior reusability, making it highly promising for practical large-scale applications. These findings provide important design guidance for various hydrogen-involving applications, offering new opportunities for the development of efficient and sustainable energy systems.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work has been supported by the National Natural Science Foundation of China (No. 21965005), Natural Science Foundation of Guangxi Province (No. 2021GXNSFAA076001), Guangxi Technology Base and Talent Subject (Nos. GUIKE AD18126001, GUIKE AD20297039), and Innovation Project of Guangxi Graduate Education (Nos. YCSW2023140, YCBZ2023062).
Supplementary material associated with this article can be found, in the online version, at doi:
M. Li, Z. Zhao, Z. Xia, et al., Angew. Chem. Int. Ed. 60 (2021) 8243–8250. doi: 10.1002/anie.202016199
Z. Gao, G. Wang, T. Lei, et al., Nat. Commun. 13 (2022) 118. doi: 10.1038/s41467-021-27785-5
S.S. Liu, Z.B. Li, C.L. Jiao, et al., Int. J. Hydrog. Energy 38 (2013) 2770–2777. doi: 10.1016/j.ijhydene.2012.11.042
C. Saka, Appl. Catal. B: Environ. 292 (2021) 120165. doi: 10.1016/j.apcatb.2021.120165
Q. Yao, X. Zhang, Z.H. Lu, Q. Xu, Coord. Chem. Rev. 493 (2023) 215302. doi: 10.1016/j.ccr.2023.215302
L. Ouyang, W. Chen, J. Liu, et al., Adv. Energy Mater. 7 (2017) 1700299. doi: 10.1002/aenm.201700299
L. Yao, X. Li, W. Peng, et al., Inorg. Chem. Front. 8 (2021) 1056–1065. doi: 10.1039/D0QI01244K
Y. Zhu, L. Ouyang, H. Zhong, et al., Angew. Chem. Int. Ed. 59 (2020) 8623–8629. doi: 10.1002/anie.201915988
H.X. Nunes, D.L. Silva, C.M. Rangel, A.M.F.R. Pinto, Energies 14 (2021) 3567. doi: 10.3390/en14123567
A.F. Baye, M.W. Abebe, R. Appiah-Ntiamoah, H. Kim, J. Colloid Interface Sci. 543 (2019) 273–284. doi: 10.1016/j.jcis.2019.02.065
H. Zhang, G. Xu, L. Zhang, et al., Renew. Energy 162 (2020) 345–354. doi: 10.1016/j.renene.2020.08.031
X. Zhang, X. Sun, D. Xu, et al., Appl. Surf. Sci. 469 (2019) 764–769. doi: 10.1016/j.apsusc.2018.11.094
C. Wu, J. Guo, J. Zhang, et al., Renew. Energy 136 (2019) 1064–1070. doi: 10.1016/j.renene.2018.09.070
X. Wang, J. Liao, H. Li, H. Wang, R. Wang, Int. J. Hydrog. Energy 42 (2017) 6646–6656. doi: 10.1016/j.ijhydene.2016.12.032
Z. Liu, Y. Tian, S. Li, et al., Adv. Funct. Mater. 33 (2023) 2301994. doi: 10.1002/adfm.202301994
H. Sun, J. Meng, L. Jiao, F. Cheng, J. Chen, Inorg. Chem. Front. 5 (2018) 760–772.
S. Mehdi, Y. Liu, H. Wei, et al., Appl. Catal. B: Environ. 325 (2023) 122317. doi: 10.1016/j.apcatb.2022.122317
Z. Xiao, Y. Wang, Y.C. Huang, et al., Energy Environ. Sci. 10 (2017) 2563–2569. doi: 10.1039/C7EE01917C
Y. Fan, Y. Zhou, L. Zhang, Y. Feng, K. Shih, Sep. Purif. Technol. 264 (2021) 118367. doi: 10.1016/j.seppur.2021.118367
G. Zhang, G. Wang, Y. Liu, et al., J. Am. Chem. Soc. 138 (2016) 14686–14693. doi: 10.1021/jacs.6b08491
J. Zhang, J. Zhang, F. He, et al., Nano Micro Lett. 13 (2021) 65. doi: 10.1007/s40820-020-00579-y
X. Wang, Y. Zhang, H. Si, et al., J. Am. Chem. Soc. 142 (2020) 4298–4308. doi: 10.1021/jacs.9b12113
W. Zhong, Z. Wang, N. Gao, et al., Angew. Chem. Int. Ed. 59 (2020) 22743–22748. doi: 10.1002/anie.202011378
Y. Shan, M. Zhang, Y. Bai, et al., Chem. Eng. J. 429 (2022) 132146. doi: 10.1016/j.cej.2021.132146
L. He, Y. Cheng, Q. Li, et al., Chem. Eng. J. 453 (2023) 139566. doi: 10.1016/j.cej.2022.139566
S. Sun, Y. Tang, C. Wu, C. Wan, Anal. Chim. Acta 1107 (2020) 55–62. doi: 10.1016/j.aca.2020.02.014
X. Wang, Z. Na, D. Yin, et al., ACS Nano 12 (2018) 12238–12246. doi: 10.1021/acsnano.8b06039
T. Chen, F. Wang, S. Cao, et al., Adv. Mater. 34 (2022) 2201779. doi: 10.1002/adma.202201779
W. Peng, S. Liu, X. Li, et al., Chin. Chem. Lett. 33 (2022) 1403–1406. doi: 10.1016/j.cclet.2021.08.033
C. Yan, Q. Ma, F. Wang, et al., Chem. Eng. J. 433 (2022) 133651. doi: 10.1016/j.cej.2021.133651
P. Wu, Q. Jia, J. He, et al., J. Hazard. Mater. 391 (2020) 122183. doi: 10.1016/j.jhazmat.2020.122183
J. Xiong, J. Luo, L. Yang, et al., J. Ind. Eng. Chem. 64 (2018) 383–389. doi: 10.1016/j.jiec.2018.04.001
D. Chen, Y. Wu, Z. Huang, J. Chen, Nano Micro Lett. 14 (2022) 156. doi: 10.1007/s40820-022-00912-7
L. Liu, J. Liu, K. Sun, et al., Chem. Eng. J. 411 (2021) 128629. doi: 10.1016/j.cej.2021.128629
D. Roy, K. Panigrahi, B.K. Das, et al., Nanoscale Adv. 3 (2021) 4739–4749. doi: 10.1039/D1NA00304F
X. Chen, P. Ye, H. Wang, et al., Adv. Funct. Mater. 33 (2023) 2212915. doi: 10.1002/adfm.202212915
Y. Yang, Y. Huang, S. Zhou, et al., J. Energy Chem. 72 (2022) 395–404. doi: 10.1016/j.jechem.2022.06.011
C. Wang, L.L. Gu, S.Y. Qiu, et al., Appl. Catal. B: Environ. 297 (2021) 120452. doi: 10.1016/j.apcatb.2021.120452
K. Zeng, W. Li, Y. Zhou, et al., Chem. Eng. J. 421 (2021) 127831. doi: 10.1016/j.cej.2020.127831
K. Xiang, D. Wu, Y. Fan, et al., Chem. Eng. J. 425 (2021) 130583. doi: 10.1016/j.cej.2021.130583
X. Qiao, H. Kang, Y. Li, et al., Appl. Catal. B: Environ. 305 (2022) 121034. doi: 10.1016/j.apcatb.2021.121034
M. Asnavandi, Y. Yin, Y. Li, C. Sun, C. Zhao, ACS Energy Lett. 3 (2018) 1515–1520. doi: 10.1021/acsenergylett.8b00696
K. Zhang, G. Zhang, J. Qu, H. Liu, Small 14 (2018) 1802760. doi: 10.1002/smll.201802760
X. Wang, C. Sun, F. He, et al., ACS Appl. Mater. Interfaces 11 (2019) 32460–32468. doi: 10.1021/acsami.9b07975
S.J. Sitler, K.S. Raja, I. Charit, J. Electrochem. Soc. 163 (2016) H1069–H1075. doi: 10.1149/2.0201613jes
W. Li, Y. Li, H. Fu, et al., Chem. Eng. J. 381 (2020) 122683. doi: 10.1016/j.cej.2019.122683
M. Zhang, Y. Liu, H. Zhao, et al., ACS Appl. Mater. Interfaces 13 (2021) 19904–19914. doi: 10.1021/acsami.0c23007
X. Hu, J. Wang, J. Wang, et al., Appl. Catal. B: Environ. 318 (2022) 121879. doi: 10.1016/j.apcatb.2022.121879
T. Zhao, D. Zhong, G. Hao, Q. Zhao, Appl. Surf. Sci. 607 (2023) 155079. doi: 10.1016/j.apsusc.2022.155079
Ö. Şahin, D.E. Karakaş, M. Kaya, C. Saka, J. Energy Inst. 90 (2017) 466–475. doi: 10.1016/j.joei.2016.03.003
P. Li, Y. Huang, Q. Huang, et al., Appl. Catal. B: Environ. 313 (2022) 121444. doi: 10.1016/j.apcatb.2022.121444
J. Li, X. Hong, Y. Wang, et al., Energy Storage Mater. 27 (2020) 187–197. doi: 10.1016/j.ensm.2020.01.011
A. Tang, C. Wan, X. Hu, X. Ju, Nano Res. 14 (2021) 4063–4072. doi: 10.1007/s12274-021-3341-z
Y. Huang, L. Zhang, L.W. Jiang, et al., Small 19 (2023) 2302970. doi: 10.1002/smll.202302970
S. Zhou, L. Cheng, Y. Huang, et al., Appl. Catal. B: Environ. 328 (2023) 122519. doi: 10.1016/j.apcatb.2023.122519
H. Li, Z. Liu, L. Wang, et al., Chem. Eur. J. 29 (2023) e202203207. doi: 10.1002/chem.202203207
F.O. Baydaroglu, E. Özdemir, A.G. Gürek, Int. J. Hydrog. Energy 47 (2022) 9643–9652. doi: 10.1016/j.ijhydene.2022.01.052
H. Zhang, L. Zhang, I.A. Rodríguez-Pérez, et al., Appl. Surf. Sci. 540 (2021) 148296. doi: 10.1016/j.apsusc.2020.148296
P. Li, R. Chen, S. Zhao, et al., Appl. Catal. B: Environ. 298 (2021) 120523. doi: 10.1016/j.apcatb.2021.120523
H. Zhang, Y. Liu, H. Wei, et al., Appl. Catal. B: Environ. 314 (2022) 121495. doi: 10.1016/j.apcatb.2022.121495
S. Zhou, Y. Yang, W. Zhang, et al., J. Colloid Interface Sci. 591 (2021) 221–228. doi: 10.1016/j.jcis.2021.02.009
H. Wu, Y. Cheng, B. Wang, et al., J. Energy Chem. 57 (2021) 198–205. doi: 10.1016/j.jechem.2020.08.051
A. Sermiagin, D. Meyerstein, G.S. Rolly, et al., Int. J. Hydrog. Energy 47 (2022) 3972–3979. doi: 10.1016/j.ijhydene.2021.11.040
U.B. Demirci, P. Miele, C.R. Chim 17 (2014) 707–716. doi: 10.1016/j.crci.2014.01.012

Figure 1 (a) Schematic diagram for the preparation of Co2B-CoPOx. (b) XRD pattern of Co2B-CoPOx. (c) EPR spectra of as-synthesized samples at different volume of PA.

Figure 2 (a) SEM image of Co2B-CoPOx. (b) TEM and (c) high-resolution TEM (HR-TEM) images of Co2B-CoPOx (inset: SADE pattern). High resolution XPS spectra of (d) Co 2p and (e) B 1s for Co2B-CoPOx and Co2B-ZIF-67. (f) P 2p and (g) O 1s for Co2B-CoPOx.

Figure 3 (a) The equivalent H2 per mole of sodium borohydride versus time with different catalysts and (b) the corresponding HGR values. (c) The equivalent H2 per mole of sodium borohydride versus time with different volume of PA and (d) the corresponding HGR values.

Figure 4 (a) Curves of hydrolysis of alkaline NaBH4 solution with different reaction temperatures in the range of 298–318 K. (b) Comparison of the HGR and Ea values of Co2B-CoPOx with other recently reported catalysts. (c) Reusability test of Co2B-CoPOx catalyst in alkaline NaBH4 solution at 25 ℃ and (d) the corresponding HGR values in the different cycle. (e) The proposed mechanism diagram of NaBH4 hydrolysis for H2 generation.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: