CAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, China
b.
Department of Neurosurgical, The First Affiliated Hospital of University of Science and Technology of China, Division of Life Sciences and Medicine, Hefei 230026, China
Received Date:
06 September 2023 Accepted Date:
15 October 2023 Revised Date:
12 October 2023 Available Online:
15 June 2024
Abstract:
Single-chain nanoparticles represent an emerging class of nanomaterials designed to mimic protein's folding paradigm. Intrachain covalent crosslinking toward the formation of single-chain nanoparticles encounters complex energy landscapes, leading to the potential occurrence of misfolding issues. While non-covalent crosslinking can circumvent this issue, the resulting single-chain nanoparticles exhibit lower structural stability compared to their covalently crosslinked counterparts. In this study, we present a novel approach for the synthesis of single-chain nanoparticles, achieved through the combination of non-covalent and covalent intramolecular crosslinking. Cyanostilbenes grafted onto the linear polymer form intrachain non-covalent stacks aided by hydrogen bonds, leading to the formation of non-covalently crosslinked single-chain nanoparticles. These nanoparticles undergo conversion to covalently crosslinked nanostructures through subsequent photo-irradiation using [2 + 2] photocycloaddition, a process facilitated by the supramolecular confinement effect. Consequently, the resulting single-chain nanoparticles demonstrate both intrachain folding efficiency and substantial stability, offering significant potential for advancing applications across diverse fields.
Protein folding yields globular tertiary structures from linear polypeptide chains, facilitating essential biological functions including transmembrane translocation, trafficking, secretion, immune responses, and cell cycle regulation [1,2]. Drawing inspiration from proteins, the folding of synthetic polymers into precisely defined single-chain nanoparticles (SCNPs) has garnered significant interest due to their potential applications in sensors, catalysis, and drug delivery [3-8]. A feasible strategy for fabricating SCNPs involves coupling and collapsing linear polymers that contain reactive cross-linking groups. To date, a diverse range of covalent chemical reactions, including isocyanate reactions, click reactions and Diels–Alder reactions [9-12], have been devised to induce the folding of linear polymers. Despite the progress made, these covalent chemical reactions feature complex energy landscapes during the folding process, leading to the creation of a sequence of partially folded states as metastable intermediates [13-19]. Accordingly, careful regulation of temperature and solvent is essential for adjusting the energy landscape and thereby preventing misfolding.
In stark contrast, natural protein folding occurs with rapidity and efficiency. Folded proteins invariably adopt the thermodynamically most stable conformation, achieved through the polypeptide chain's spontaneous exploration of its lowest-energy structure via a trial-and-error process [20-22]. The intra-chain folding of synthetic polymers propelled by non-covalent crosslinking is inherently spontaneous, dictated by their lowest energy state, analogous to protein folding [23-25]. Nevertheless, these non-covalently crosslinked SCNPs suffer from lower structural stability than their covalent counterparts. Their susceptible to environmental factors poses constraints on their practical utility [26,27]. We hypothesize that SCNPs driven by non-covalent crosslinking, when equipped with reaction sites, can promote subsequent covalent fixation through a supramolecular confinement effect [28-32]. The resultant SCNPs concurrently feature high folding efficiency and adequate stability, thereby offering advantages over either covalent or non-covalent bonds-driven SCNPs reported in existing literatures.
Taking this into consideration, herein we sought to fabricate SCNPs by merging covalent and non-covalent intrachain crosslinking strategies. Specifically, a random copolymer 1 (Scheme 1) has been designed with the incorporation of π-conjugated cyanostilbene into the side chain. Two amides are attached to both ends of the cyanostilbene rod, which facilitate non-covalent π–π stacking of cyanostilbenes aided by hydrogen bonding interactions. It leads to the formation of SCNPs in apolar solvents through non-covalent crosslinking. The resulting SCNPs offer a confined space with a short center-to-center distance between neighboring cyanostilbenes (<4.2 Å for the olefin units) [33]. In view of the photo-reactive character of cyanostilbenes, complete [2 + 2] photo-cycloaddition (Scheme 1) takes place upon exposure to light, causing the transition of SCNPs from non-covalent to covalent crosslinking. This process enhances the stability of SCNPs, as manifested by their capability to maintain nanoparticle morphology even in polar solvents. Overall, the current study provides a novel and efficient approach to fabricate SCNPs.
Scheme 1
Scheme 1.
Schematic representation for collapsing polymer 1 into single chain nanoparticles in apolar media by merging non-covalent and covalent intramolecular crosslinking. The chemical structure of polymer 1, along with its synthetic procedure, are shown in the frame.
The targeted polymer 1 was synthesized through ring-opening metathesis copolymerization, utilizing monomers 2 and 3 (Scheme 1) at a 1:3 feeding ratio with the third-generation Grubbs catalyst. Through monitoring via1H NMR spectra, the norbornene resonance peak, initially at 6.2–5.9 ppm, vanished after an 8-h reaction, signifying successful polymerization (Fig. S1 in Supporting information). The cyanostilbene unit’s mole percentage in copolymer 1 was calculated to be 21.7% (Fig. S2 in Supporting information), akin to the monomer feed ratio, by utilizing the 1H NMR integration ratio of the aromatic peak Ha (6.98 ppm) and the alkyl chain peaks (0.87 and 4.00 ppm). Size exclusion chromatography (SEC) determined the molecular weight of 1 as 58.2 kDa, accompanied by a dispersity index (PDI) value of 1.17.
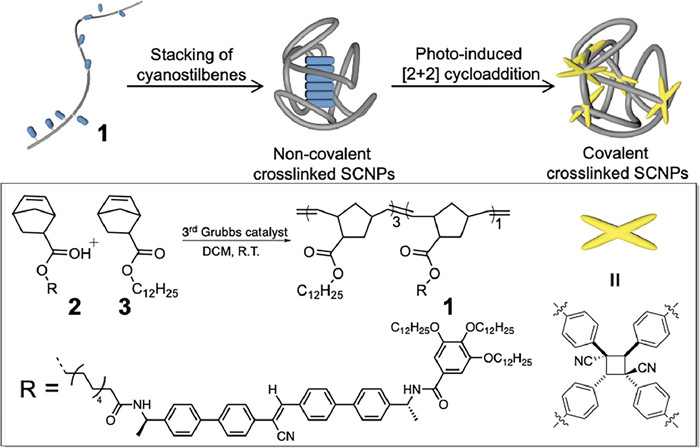
Non-covalent stacking of cyanostilbenes was firstly studied through absorption and emission spectra in different solvents. As widely documented, the molecularly dissolved state prevails for π-conjugated systems in polar solvents such as chloroform and tetrahydrofuran (THF) (“good” solvents), while aliphatic solvents like MCH (methylcyclohexane, “bad” solvents) facilitate hydrogen bonding and π–π stacking [34,35]. Upon changing the solvent from chloroform to MCH, 1 exhibited a notable blue shift in the absorption band of the cyanostilbene's π–π* transition from 356 nm to 332 nm (Fig. 1a). A strong fluorescent emission centered at 492 nm emerged in MCH, distinct from the negligible emission observed in chloroform, thereby confirming the aggregation-induced emission enhancement due to the stacking of cyanostilbene units (Fig. S3c in Supporting information) [36-41]. Similar absorption and emission signal changes occurred for the precursor compound 2 at the same cyanostilbene concentration (Figs. S3a and b in Supporting information), indicating the unaltered non-covalent stacking of cyanostilbene by the polynorbornene chain in 1.
Figure 1
Figure 1.
(a) Absorption and (b) CD spectra of 1 in chloroform (black lines) and MCH (red lines) at 298 K. (c) Normalized CD intensities of polymer 1 (red line) and the precursor monomer 2 (blue line) at 348 nm by switching the volume ratio of MCH in chloroform. (d) Normalized CD intensities of polymer 1 (red line) and the precursor monomer 2 (blue line) at 348 nm by varying the temperature. All of the measured solutions were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L.
Circular dichroism (CD) spectroscopy is a powerful technique for evaluating the stacked nanostructure's regularity [42]. On this account, two stereogenic N-[(1R)-phenylethyl] benzamides (Scheme 1) are attached neighbouring to the cyanostilbene core, capable of introducing helical bias during non-covalent stacking. No Cotton effects existed for 1 in the molecularly dissolved state (dissolved in chloroform or THF, Fig. 1b). In comparison, 1 exhibited Cotton effects in MCH, displaying positive maxima at 319 nm and negative maxima at 348 nm (gabs value = 3.9 × 10−3 for 1). This observation signifies the transfer of chirality from enantiomerically pure N-[(1R)-phenylethyl]benzamides at the molecular level to the cyanostilbene core at the supramolecular level [43]. The emergence of supramolecular chirality relies on the two-fold intermolecular hydrogen bonds between adjacent cyanostilbenes, evident by the negligible Cotton effect observed in the counterpart compound with the methylated amides (Fig. S4 in Supporting information).
CD experiments were further conducted in a chloroform/MCH mixture, with a gradual increase in chloroform content in MCH. Notably, for the precursor monomer 2, CD signals exhibited remarkable sensitivity to solvent polarity variations. A mere 10% addition of chloroform (CHCl3:MCH = 1:9, v/v) resulted in the complete disappearance of the CD signal, supporting the breakup of the non-covalent cyanostilbene stacks (Fig. 1c and Fig. S5 in Supporting information). In contrast, 1 displayed a robust CD signal under identical conditions. It gradually diminished with increasing chloroform content and ultimately vanished at a CHCl3/MCH volume ratio of 1:1 (v/v, Fig. 1c). As a result, the non-covalent stacked nanostructures of 1 exhibited enhanced stability towards changes in solvent polarity in comparison to 2.
Temperature-dependent CD melting curves were obtained for both polymer 1 and the precursor monomer 2 in MCH, enabling a thorough comparison of their supramolecular stacking behaviours. A ramping rate of 1 K/min was applied to ensure controlled disassembly of the cyanostilbene stacks under thermodynamic conditions. Compound 2 exhibited a non-sigmoidal melting curve (Fig. 1d), indicative of the cooperative nucleation–elongation mechanism adopted in the non-covalent stacking process [44]. The onset temperatures of elongation increased with higher monomer concentrations (306 K at 10 µmol/L; 316 K at 20 µmol/L; and 320 K at 30 µmol/L, Fig. S6a in Supporting information), providing evidence for intermolecular stacking [5] in 2. In contrast, the melting curves of polymer 1 displayed neither sigmoidal nor non-sigmoidal characteristics (Fig. S6b in Supporting information). Additionally, the melting temperature of 1 remained constant at 336 K, despite varying cyanostilbene concentrations from 10 µmol/L to 30 µmol/L. While the melting temperature is unrelated to the overall cyanostilbene concentration, it closely correlates with the local concentration. This is substantiated by the decrease in melting temperature for copolymers with reduced percentages of cyanostilbene units (Tm: 333 K for 22.9% percentage of cyanostilbene unit, 323 K for 12.2% percentage of cyanostilbene unit, 308 K for 3.2% percentage of cyanostilbene unit; Fig. S6d in Supporting information). These characteristics in the melting curves bear resemblance to those obtained during the thermal denaturation of intramolecularly folded proteins and peptides [5,45]. In conclusion, this validates the intra-chain non-covalent stacking of cyanostilbenes within polymer 1.
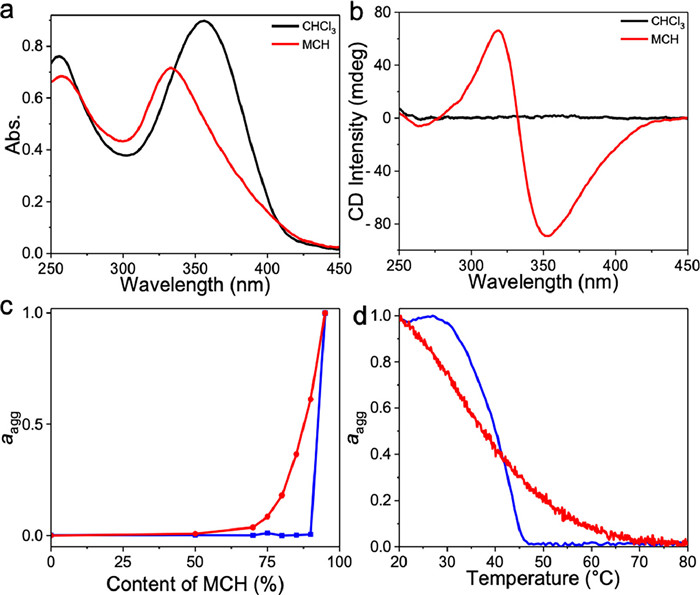
Furthermore, the morphologies of polymer 1 and the precursor monomer 2 upon non-covalent cyanostilbene stacking were compared. The directional non-covalent forces contributing to intermolecular stacks in 2 lead to anisotropic growth. Consequently, this led to nanofibers which tend to intertwine with each other (Fig. S8 in Supporting information). In contrast, the presence of the polynorbornene main chain in 1 restricts one-dimensional stacking of cyanostilbenes. Intra-chain stacking of cyanostilbenes enables collapse of linear polymer 1 in “good” solvents into SCNPs in “bad” solvents. This conclusion was substantiated through AFM measurements, revealing the formation of nanoparticles with a height of approximately 6 nm and the diameter of about 65 nm (Fig. 2a and Fig. S9 in Supporting information). It is worth noting that the observed diameter in AFM (65 nm) exceeded that in dynamic light scattering (DLS, approximately 8 nm, Fig. 2b). This discrepancy, also encountered in the previous SCNPs systems, is primarily attributed to solvent evaporation during the AFM sample preparation process, leading to the aggregation of SCNPs [9,46,47].
Figure 2
Figure 2.
(a) AFM image on mica, and (b) DLS data of 1 in MCH. The measured samples were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L.
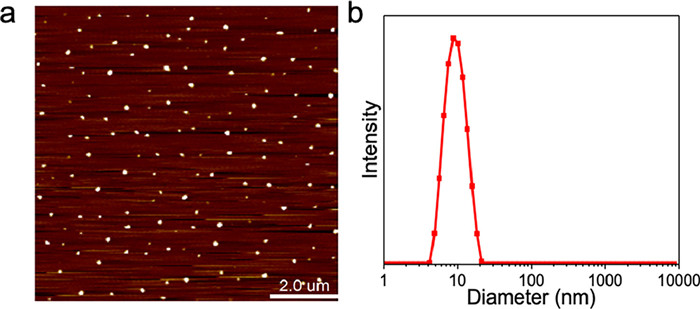
Subsequently, we focused on converting the SCNPs derived from 1via non-covalent to covalent crosslinking, achieved by conducting a photo-irradiated reaction of cyanostilbenes. In MCH, the π–π* absorption peak around 332 nm vanished after 5 min of 430 nm (12 W) light irradiation, giving rise to a new absorption band at 267 nm (Fig. 3a). Simultaneously, the Cotton effect of cyanostilbenes diminished, leading to a subtle Cotton effect below the 320 nm wavelength (Fig. S11a in Supporting information). In contrast, photo-illumination in “good” solvents like THF induced a slight blue shift in the π–π* absorption band from 356 nm to 336 nm in the photo-stationary state (Fig. 3b). No new absorption band appeared in the low-energy absorption region. The findings suggest the formation of distinct photo-products in “good” and “bad” solvents.
Figure 3
Figure 3.
(a) UV–vis spectra of polymer 1 in the "bad" solvent MCH upon irradiation with 430 nm LED lamp (12 W) for 300 s. (b) UV–vis spectra of polymer 1 in the “good” solvent THF upon irradiation with 430 nm lamp for 20 s. All of the measured solutions were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L. (c) Structures of the photo-products after illumination of the cyanostilbenes in 1 or 2 in the stacked state and monomeric state.
We then aimed to characterize the two distinct photo-products of cyanostilbene. The poorly resolved signals and polydisperse structural nature of compound 1 hinder the determination of photo-products using 1H NMR and MALDI-TOF techniques. To address this, we utilized compound 2 as the control sample; it exhibited analogous spectroscopic alterations upon light irradiation in both “good” and “bad” solvents (Figs. S11b and c in Supporting information). The 1H NMR of the photo-product in THF revealed a marginal shift of the cyano-vinyl protons from 7.6 ppm to 7.4 ppm. It supports the configuration isomerization with the Z/E ratio of 19.5:80.5 at the photo-stationary state (Fig. 3c and Fig. S12 in Supporting information). The MALDI-TOF data remained unchanged throughout the process (1429.9, Fig. S13 in Supporting information), confirming the intramolecular Z/E transformation of the cyanostilbene unit. In contrast, a noticeable shift of the cyano-vinyl resonances to 5.3 ppm was observed for the photo-product of 2 in MCH (Fig. S13). This indicates that the cyano-vinyl group lost its aromatic character, resulting in the formation of aliphatic cyclobutane rings. Additionally, the photo-product exhibited a nearly two-fold increase in mass spectra (2855.1, Fig. S13). The findings signify the occurrence of [2 + 2] photocycloaddition of cyanostilbene in MCH (Fig. 3c). It is rationalized that non-covalent stacking in “bad” solvents provides a confined space, resulting in a short center-to-center distance between adjacent cyanostilbenes. This leads to a distinct photo-illuminated product for cyanostilbenes from that in “good” solvents.
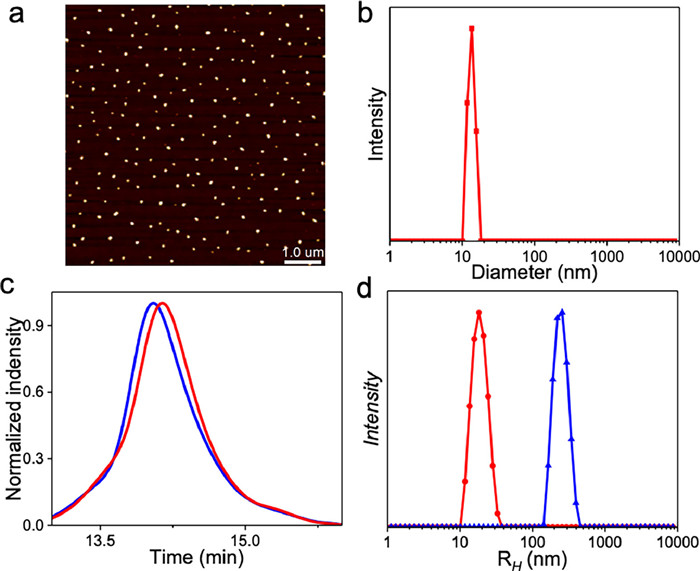
The [2 + 2] photocycloaddition of cyanostilbenes within 1 enables intra-chain covalent crosslinking of SCNPs in MCH. This conclusion is verified through AFM, which revealed the formation of nanoparticles of comparable size to that of non-covalently crosslinked SCNPs before light irradiation (Fig. 4a). The hydrodynamic diameter of SCNPs after photo-illumination was measured at 12 nm using DLS (Fig. 4b), slightly larger than that of non-covalently crosslinked SCNPs (8 nm). The formation of covalently crosslinked SCNPs was additionally substantiated via SEC (size exclusion chromatography), revealing a longer elution time compared to the unfolded polymer 1prior to light irradiation (14.0 min versus 14.2 min, Fig. 4c). Conversely, photo-irradiation of linear polymer 1 in THF did not result in SCNPs formation due to the Z/E transformation of the cyanostilbene units. This is evident from the absence of nanoparticles in AFM, together with the observation of a substantial hydration radius using DLS (255 nm, Fig. S14 in Supporting information).
Figure 4
Figure 4.
(a) AFM image of 1 on mica surface after 430 nm photo-irradiation in MCH. (b) DLS data of 1 in MCH after 430 nm photo-irradiation in MCH. (c) SEC spectra (eluent: THF) of 1 before and after photo-irradiation in MCH (blue and red lines, respectively). (d) DLS data of linear polymer 1 in THF (blue line), along with collapsing 1 upon photo-irradiation in MCH to form covalently crosslinked SCNPs, followed by evaporation the MCH solution and redissolved in THF (red line). All of the measured solutions were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L.
Finally, the stability of SCNPs was examined before and after light irradiation. The MCH solvent used for SCNPs formation was evaporated, and polar solvent THF was then added. In the absence of light irradiation, the non-covalently crosslinked SCNPs disintegrated in THF. This was confirmed by a notable rise in hydrodynamic diameter as observed in DLS (217 nm, Fig. 4d), along with the restoration of cyanostilbene’s absorption to its molecularly dissolved state. In contrast, the covalently crosslinked SCNPs remained stable in THF, maintaining a hydrodynamic diameter of 18 nm in DLS (Fig. 4d). The measured size is relatively larger in THF compared to MCH (12 nm), a reasonable outcome attributed to the deswelling effect in polar media. Thus, the covalently crosslinked SCNPs via photo-irradiated [2 + 2] photo-cycloaddition of cyanostilbenes exhibit enhanced stability in polar solvents than the non-covalently crosslinked counterparts.
In summary, we have developed a novel strategy for forming SCNPs by combining non-covalent and covalent intramolecular crosslinking. The fundamental design principle involves grafting photo-reactive cyanostilbenes onto linear polymer 1. Cyanostilbenes tend to form intrachain non-covalent stacks in MCH, facilitated by hydrogen bonding and π-π stacking interactions, resulting in the formation of non-covalently crosslinked SCNPs. This local stacking confines the cyanostilbenes, leading to the quantitative formation of [2 + 2] photocycloaddition products upon exposure to 430 nm light. Consequently, this transformation converts non-covalently crosslinked SCNPs into covalently crosslinked ones, characterized by robust stability even in polar solvents. The resulting SCNPs exhibit both high folding efficiency due to their non-covalent crosslinked nature and sufficient stability thanks to post-covalent fixation. This combination of advantages avoids the drawbacks associated with either covalent or non-covalent bond-driven SCNPs reported in existing literature, holding promise for advancing SCNPs' applications across various fields.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 22371272 and 22301295), the Fundamental Research Funds for the Central Universities (Nos. YD2060002036 and WK5290000004), International Partnership Program of the Chinese Academy of Sciences (No. 123GJHZ2022064MI), and the Collaborative Innovation Program of Hefei Science Center, CAS (No. 2022HSC-CIP014).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109217.
R. Liao, F. Wang, Y. Guo, Y. Han, F. Wang, J. Am. Chem. Soc. 144 (2022) 9775–9784. doi: 10.1021/jacs.2c02270
[44]
F. Wang, R. Liao, F. Wang, Angew. Chem. Int. Ed. 62 (2023) e202305827.
[45]
P.L. Privalov, Temperature-induced changes in protein, in: C.B. Anfinsen, J.T. Edsall, F.M. Richards (Eds.), Advances in Protein Chemistry, Academic Press, New York, 1979, pp. 167–241.
T. Dünnebacke, K.K. Kartha, J.M. Wahl, R.Q. Albuquerque, G. Fernández, Chem. Sci. 11 (2020) 10405–10413. doi: 10.1039/d0sc03442h
Scheme 1
Schematic representation for collapsing polymer 1 into single chain nanoparticles in apolar media by merging non-covalent and covalent intramolecular crosslinking. The chemical structure of polymer 1, along with its synthetic procedure, are shown in the frame.
Figure 1
(a) Absorption and (b) CD spectra of 1 in chloroform (black lines) and MCH (red lines) at 298 K. (c) Normalized CD intensities of polymer 1 (red line) and the precursor monomer 2 (blue line) at 348 nm by switching the volume ratio of MCH in chloroform. (d) Normalized CD intensities of polymer 1 (red line) and the precursor monomer 2 (blue line) at 348 nm by varying the temperature. All of the measured solutions were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L.
Figure 2
(a) AFM image on mica, and (b) DLS data of 1 in MCH. The measured samples were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L.
Figure 3
(a) UV–vis spectra of polymer 1 in the "bad" solvent MCH upon irradiation with 430 nm LED lamp (12 W) for 300 s. (b) UV–vis spectra of polymer 1 in the “good” solvent THF upon irradiation with 430 nm lamp for 20 s. All of the measured solutions were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L. (c) Structures of the photo-products after illumination of the cyanostilbenes in 1 or 2 in the stacked state and monomeric state.
Figure 4
(a) AFM image of 1 on mica surface after 430 nm photo-irradiation in MCH. (b) DLS data of 1 in MCH after 430 nm photo-irradiation in MCH. (c) SEC spectra (eluent: THF) of 1 before and after photo-irradiation in MCH (blue and red lines, respectively). (d) DLS data of linear polymer 1 in THF (blue line), along with collapsing 1 upon photo-irradiation in MCH to form covalently crosslinked SCNPs, followed by evaporation the MCH solution and redissolved in THF (red line). All of the measured solutions were kept in the same cyanostilbene concentration of 2 × 10−5 mol/L.

 DownLoad:
DownLoad:

 下载:
下载:






