
Scheme 1.
Background and concept of the work.
Asymmetric alkenylation reaction of benzoxazinones with diarylethylenes catalyzed by B(C6F5)3/chiral phosphoric acid
Zhen Liu , Zhi-Yuan Ren , Chen Yang , Xiangyi Shao , Li Chen , Xin Li

Stereochemically defined alkenes are prominent structures in natural products, pharmaceutical ingredients and bioactive compounds [1,2]. In addition, they are important building blocks in asymmetric synthesis because the alkene group can be functionalized in many ways. Consequently, efficient catalytic methods toward the synthesis of chiral alkenes with high enantiopurity has long been pursued in synthetic organic chemistry [3]. Catalytic asymmetric alkenylation, which alkenyl reagents are asymmetrically added to unsaturated bonds or stereoselectively cross-coupled with alkyl halides, is considered as one of the most efficient method to construct chiral alkene molecules [4-7]. Asymmetric alkenylation of traditional alkenyl reagents, which is heavily dependent on alkenyl metals [8-10] or metalloids [11-20], has been widely developed (Scheme 1a). The use of these traditional alkenyl reagents usually produces waste such as metal ions, which does not meet the requirements of green chemistry. Therefore, non-metal-based alkenes are considered to be ideal alkenyl reagents. However, the use of non-metal-based alkenes as alkenyl reagents is less explored. In 1989, the first asymmetric Heck reaction was published by using palladium catalyst which realized the stereoselective cross-coupling of alkenes [21-23]. Since then, the work of asymmetric alkenylation of non-metal-based alkenes by organometallic catalysis has been continuously developing [24-29]. In 2015, Jia developed the enantioselective addition reaction of N-sulfonyl α-ketiminoesters and styrenes catalyzed by Nickel catalyst [30,31]. In addition, compared to metal catalysts, metal-free catalysts have greater limitations, making it more difficult to catalyze the reaction of non-metal-based alkenes. Therefore, very limited approached have been disclosed for the asymmetric alkenylation by metal-free catalysis. The alkenyl reagents used in previously reported examples catalyzed by metal-free catalysts are either alkenes containing electron rich indole groups that make them more nucleophilic, or the alkenes containing activated groups such as phenolic hydroxyl groups [32-36]. So the development of efficient strategy, especially by metal-free catalysis, for the asymmetric alkenylation with simple alkenes, is very meaningful and highly desirable.
Chiral benzoxazines and benzoxazinones constitute the key organic scaffolds of various active pharmaceuticals and agricultural chemicals (Scheme 1b) [37-40]. Over the past several decades, the development of approaches for the synthesis of these compounds has attracted considerable attention [41-46]. However, there is only one example of asymmetric alkenylation of benzoxazinones catalyzed by [Rh(COE)2Cl]2 with a chiral sulfur-olefin ligand, which suffered from poor reactivity and harsh reaction conditions [42]. Accordingly, it is extremely significant to develop an efficient asymmetric alkenylation method of benzoxazinone.
On the other hand, the use of simple alkenes as alkenyl reagents is inexpensive and atom economic, but there exist several important challenges. First, simple alkenes are relatively stable, which are difficult to be activated by metal-free catalysts. Second, because simple alkenes have no activated site that can form strong interactions with the catalyst, it is not easy to control the stereoselectivity. Originally pioneered by Ishihara, the combination of boron type Lewis acid and chiral phosphoric acids (CPAs) significantly improves the activity of the catalyst and the selectivity of the reaction [47]. Very recently, we have realized the enantioselective ene reaction and aza-Diels-Alder reaction of ketimine by a B(C6F5)3/CPA catalyst system, and found that the imine substrate can be sufficiently activated by B(C6F5)3/CPA system to react with simple alkenes [48,49]. Based on this finding, we surmised that the B(C6F5)3/CPA system can also reduce the LUMO orbital energy of benzoxazinone and match it with the HOMO orbital energy of proper simple alkenes. Herein, we report an efficient asymmetric alkenylation of benzoxazinones with unactivated diarylethylenes under mild conditions through B(C6F5)3/CPA catalyst system (Scheme 1c). This process does not require any sacrificial reagent and is byproduct-free. The Lewis acid enhanced Brønsted acid catalytic system effectively reduces the activation free energy barrier for the alkenylation, thus enabling an unprecedented C(sp2)−C(sp2) formation to form an alkenylated tertiary center.
To verify our hypothesis, by using 2H-benzo[b][1,4]oxazin-2-one 1a and 1,1-diphenylethylene 2a as the model substrates, we started a preliminary attempt with 2 mol% chiral phosphoric acid (S)-4a as catalyst in CH2Cl2 (DCM) at room temperature. However, only trace amounts of target product 3aa was obtained, which is obviously unsatisfactory (Table 1, entry 1). We expect that the use of boron Lewis acids in association with chiral phosphoric acid as catalyst system can enhance the outcome of this reaction. To our delight, with the addition of Lewis acid, the yield and enantioselectivity of product 3aa are increased (Table 1, entries 2–5). Especially when the catalyst system B(C6F5)3/(S)-4a is used, providing 3aa in 90% yield and 92:8 e.r. (Table 1, entries 5). Some metal type Lewis acids were also attempted,but no better results were acquired (Table S1 in Supporting information for details). Then a series of CPAs [50-53] were further evaluated, showed that 9-anthryl substituted (S)-4a was still the best one (Table 1, entries 5–7). Other kinds of CPAs were also examined with no better results obtained (Table S1). After that, the effect of solvent was investigated (Table 1, entries 8–10). It was found that the solvent had a significant impact on the yield and enantioselectivity, in which the initial used CH2Cl2 gave the best outcome (Table 1, entry 5). Fortunately, the addition of 5 Å molecular sieves could accelerate the reaction rate and increase the enantioselectivity (Table 1, entry 11). In addition, lowering the reaction temperature to −20 ℃ associated with increasing the concentration to 0.2 mol/L further raised the enantiomeric ratio value to 97:3 (Table 1, entry 12). The absolute configuration of 3aa was determined to be (R) by X-ray diffraction analysis, and those of other products were assigned by analogy.
With the optimal conditions in hand, we explored the scope of this asymmetric alkenylation reaction. As exhibited in Scheme 2, a range of substrates bearing different electronic features of the substituents (F, Cl, Br and Me) at the C7 and C8 position were well tolerated to deliver the corresponding. products 3ba-3ia in 80%–91% yields and 93:7–97.5:2.5 e.r. In addition, the benzoxazinone bearing methoxy group at C7 position, which has relatively weak electrophilicity, can also be applied to current studied catalytic strategy, afforded product 3ja in 48% yield with 95:5 e.r. (3ja) at room temperature. Similarly, the ester group at the C7 position can also achieve good results (3ka, 88:12 e.r.). Furthermore, substrates with C5 and C6 position substituents on the phenyl ring were also examined. As a result, various alkyl substituents at C5 position, such as methyl group, ethyl group and tert‑butyl group, can react smoothly regardless of steric hindrance, providing 3la–3na in 72%–84% yields and 94:6–95.5:4.5 e.r. By using CPA (S)-5b, substrates with the methyl group at C5 position and the fluoro group at C5 and C6 position could also give the desired product in 83%–89% yields and 89.5:10.5–92.5:7.5 e.r. (3oa–3qa). Moreover, disubstituted substrates had good compatibility in this B(C6F5)3/CPA catalyzed reaction, affording the corresponding products 3ra and 3sa in 70% and 80% yields with 95:5 and 88:12 e.r., respectively.

To expand the substrate scope, we also examined a range of 1,1-diarylethylene derivatives. As shown in Scheme 3, 1,1-diphenylethylenes bearing either electron-donating group (CH3 and tBu) or electron-withdrawing group (F, Cl and Br) are all tolerated to acquire the desired products in 65%–86% yields and 93.5:6.5–97:3 e.r. (3ab–3af). Furthermore, monosubstituted 1,1-diphenylethylenes 2g–2i were also investigated, in which corresponding products 3ag–3ai were obtained in good yields (64%–85%) with moderate Z/E ratios (1:1.7–1:1.9) and excellent enantioselectivities (Z: 95:5–96.5:3.5 e.r.; E: 96:4–97.5:2.5 e.r.). In addition, when 2-(1-phenylvinyl) naphthalene is used as the alkenyl reagent, the desired product 3aj can also be obtained in 90% yield with 1:1.3 Z/E and 94:6/95:5 e.r. The relative configurations of Z/E of 3ah were determined by the 2D-NOESY experiment (see Supporting information) and those of other products of 3ag, 3ai and 3aj were assigned by analogy.

To confirm the efficiency of this method in preparative synthesis, a gram-scale reaction was performed with 2H-benzo[b][1,4]oxazin-2-one 1a (5 mmol) and 1,1-diphenylethylene 2a (6 mmol). Encouragingly, the reaction gave the desired product 3aa in 83% yield and 97:3 e.r. (Scheme 4a). In order to show the applicability of this reaction, some transformations of 3aa were also studied. As shown in Scheme 4b, the alkenylation product 3aa can be easily reduced to 4 in 95% yield with maintained 97:3 e.r. in 5 h. When methanol is used as the reducing reaction solvent, a chiral amino acid derivative 5 can be obtained in 80% yield without the loss of optical purity. In addition, 3aa can be reduced by LiAlH4 at 0 ℃, and then the corresponding product 6 is obtained in 87% yield and 96:4 e.r. through the Mitsunobu reaction. Moreover, 3aa can undergo the nucleophilic attack of benzylamine and open the ring to form aryl glycine amide 7 in 84% yield and 94.5:5.5 e.r.

In order to demonstrate the mechanism, we performed several experiments. First, control experiments were conducted to investigate the cooperative effect of B(C6F5)3/(S)-4a of the alkenylation reaction. As shown in Scheme 5a, the reaction did not work under the standard reaction conditions with the absence of B(C6F5)3 or (S)-4a, indicating that B(C6F5)3 complexed with (S)-4a could greatly enhanced the reactivity of the alkenylation process. Second, we did the examination of ratio of B(C6F5)3/(S)-4a. The results showed that increasing the amount of B(C6F5)3 can accelerate the reaction without changing the stereoselectivity of the product, but increasing the amount of (S)-4a basically has no other effect on the reaction (Scheme 5b). Moreover, the nonlinear effect experiment indicated that only one chiral phosphoric acid participated in the enantiodetermining transition state (Scheme 5c).
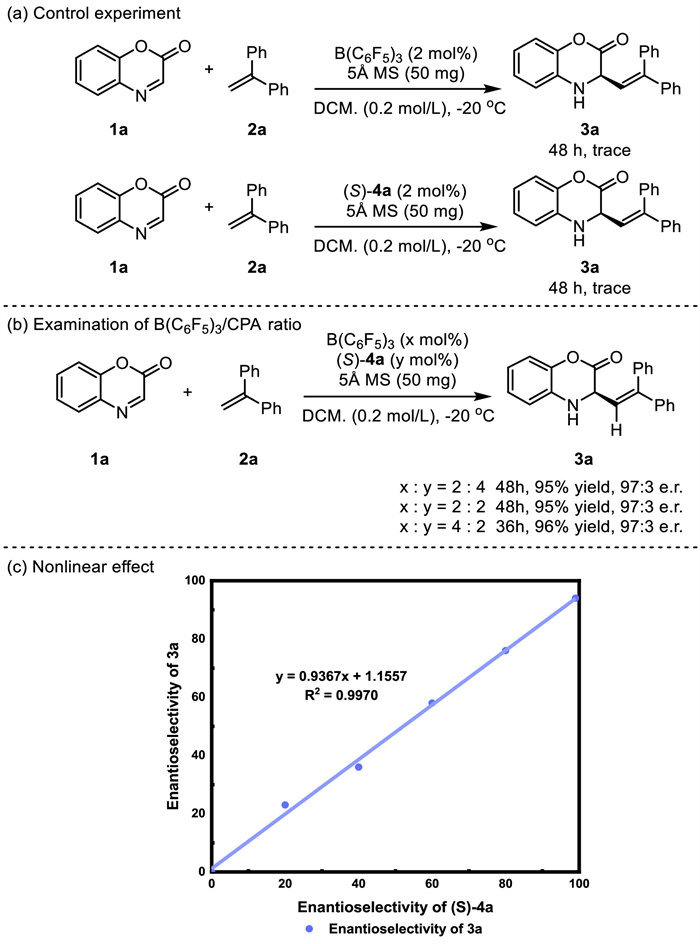
To gain insight of the origin of stereoselectivity in current studied asymmetric alkenylation reaction, a DFT calculation was conducted at the IEFPCM(CH2Cl2)/M06-2X/6-311+G(2d,p)//IEFPCM(CH2Cl2)/B3LYP/def2-SVP level of theory [54-58]. In our previous studies, a B(C6F5)3/CPA complex was proposed as the catalyst. So, we first investigated the reaction in this catalytic scenario (Scheme 6a, pathway B). The present reaction is initiated from the protonation of 1a, and the corresponding intermediate INT2-B is located at −3.2 kcal/mol. Through INT3-B, the transition states for C—C bond formation (TS1B-R & TS1B-S) are reached. The energy barrier of the transition state leading to the major enantiomer (TS1B-R) is 11.1 kcal/mol, suggesting that the reaction can occur smoothly under standard conditions. We wondered if a second B(C6F5)3 molecule could further reduce the energy barrier of the reaction by lowering the LUMO orbital energy of 1a, considering it has a carbonyl Lewis base site besides the nitrogen site. When introducing the second B(C6F5)3 molecule, a more stable intermediate INT2-A occurred, located below INT2-B by 2.4 kcal/mol. As a result, the predicted barrier of TS1A-R in the two-B(C6F5)3-involved catalytic scenario (Scheme 6a, pathway A) is only 7.7 kcal/mol, which is 3.4 kcal/mol lower than that of TS1B-R. Charge analysis of the reaction site of 1a (Fig. S1 in Supporting information) further supports this hypothesis. The Hirshfeld charge (0.80) of the reaction site (C9) in TS1A-R is 0.05 higher than that in TS1B-R, reflecting it's more electrophilic [59]. The role of B(C6F5)3, which is perhaps more important than we previously realized, is not only enhancing the acidity of CPA but also activating the benzoxazinone substrate. The favored transition state (TS1B-R) in the B(C6F5)3/CPA complex catalytic scenario is located 3.4 kcal/mol above the TS1A-R, reflecting that the B(C6F5)3/CPA complex catalytic scenario contributes little to the enantioselectivity.
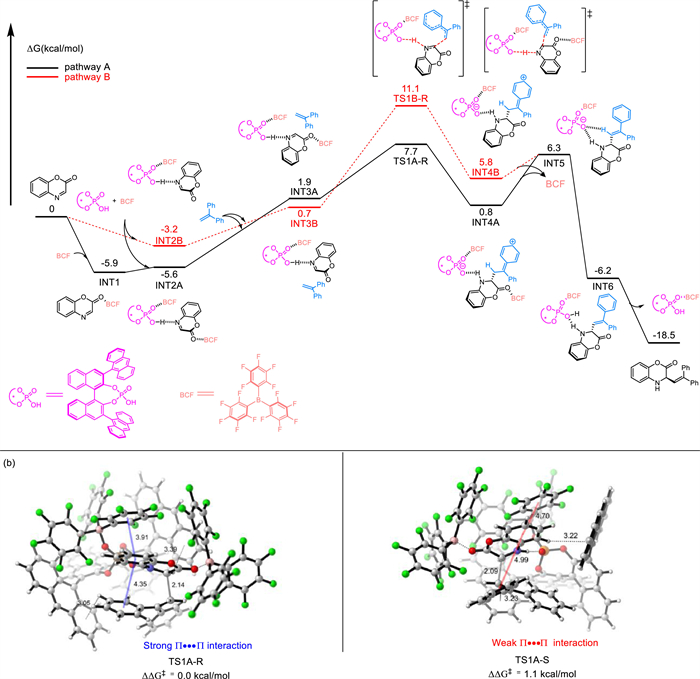
Next, we focused on the favorable catalytic scenario involving two B(C6F5)3 molecules to interpret the enantioselectivity. The energy difference between TS1A-R and TS1A-S is 1.1 kcal/mol (Scheme 6b), indicating an 90% ee value of the R enantiomer, which agrees well with the observed 94% ee value (Table 1, entry 12). Non-covalent interaction plots reveal the presence of multiple noncovalent interactions, including hydrogen bonding, C—H···π, and π···π stacking interactions in both TS1A-R and TS1A-S (Fig. S2 in Supporting information), making it difficult to determine which weak interaction dominates the enantioselectivity. Therefore, we conducted a quantitative analysis of the strength of hydrogen bonds according to electron density at the bond critical point [60]. As shown in Table S1 (Supporting information), the N—H···O hydrogen bond contributed most to stabilize the corresponding transition states among all the hydrogen bonds. The energy of this type of hydrogen bond is −9.09 kcal/mol in TS1A-R and −11.08 kcal/mol in TS1A-S, respectively. This is in line with the distance (N8—H1···O77 1.68 Å vs. N123H116···O34 1.62 Å). Although the N—H···O hydrogen bond in TS1A-R is somewhat weaker than that in TS1A-S, the presence of several other hydrogen bonds in TS1A-R may compensate for the unfavorable energy difference caused by the N—H···O hydrogen bond. Overall, these interactions make TS1A-R more stable than TS1A-S by 0.56 kcal/mol. Additionally, we observed that a sandwich structure stacking by a perfluoro phenyl group of B(C6F5)3, benzoxazinone, and diphenylethylene facilitates π···π interactions (Fig. S3 in Supporting information), lending extra help to stabilize TS1A-R. Therefore, we think that the observed enantioselectivity can be attributed to the multiple hydrogen bonding and π···π interactions.
In summary, we have achieved the enantioselective alkenylation of benzoxazinones with diarylethylenes to construct chiral alkene molecules by a simple B(C6F5)3/CPA catalyst system. Using this mild method, a wide array of benzoxazinones and diarylethylenes with various groups were well compatible in good yields with high to excellent enantioselectivities. In addition, the synthetic utility was proved by the scaled-up reaction and transformations of the products. Preliminarily mechanism was studied by control reactions, nonlinear effect experiment and theoretical calculations. Moreover, DFT studies shown that B(C6F5)3 not only increased the acidity of chiral phosphoric acid but also interacted with the substrate to activate the imine.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful to the National Natural Science Foundation of China (Nos. 22193011, 21971120 and 21933008) and National Science & Technology Fundamental Resource Investigation Program of China (No. 2018FY201200) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
M.R. Wilson, R.E. Taylor, Angew. Chem. Int. Ed. 52 (2013) 4078–4087. doi: 10.1002/anie.201207712
D. Hughes, P. Wheeler, D. Ene, Org. Process Res. Dev. 21 (2017) 1938–1962. doi: 10.1021/acs.oprd.7b00319
J. Wang, Stereoselective Alkene Synthesis, Springer, Berlin, 2012.
T. Hayashi, J. Organomet. Chem. 653 (2002) 41–45. doi: 10.1016/S0022-328X(02)01272-X
D. Müller, A. Alexakis, Chem. Commun. 48 (2012) 12037–12049. doi: 10.1039/c2cc34607a
S. Roscales, A.G. Csákÿ, Chem. Soc. Rev. 43 (2014) 8215–8225. doi: 10.1039/C4CS00195H
M.Z. Lu, J. Goh, M. Maraswami, et al., Chem. Rev. 122 (2022) 17479–17646. doi: 10.1021/acs.chemrev.2c00032
K.C. Nicolaou, W. Tang, P. Dagneau, R. Faraoni, Angew. Chem. Int. Ed. 44 (2005) 3874–3879. doi: 10.1002/anie.200500789
B. Maliszewski, T. Bauer, Adv. Synth. Catal. 361 (2019) 3689–3693. doi: 10.1002/adsc.201900363
L. Wu, H.L. Wei, J.F. Shen, J.Z. Chen, W.B. Zhang, Acta Chim. Sin. 79 (2021) 1331–1344. doi: 10.6023/a21070338
J.M. Stevens, D.W.C. MacMillan, J. Am. Chem. Soc. 135 (2013) 11756–11759. doi: 10.1021/ja406356c
X. Liu, Z. Meng, C. Li, H. Lou, L. Liu, Angew. Chem. Int. Ed. 54 (2015) 6012–6015. doi: 10.1002/anie.201500703
Q.F. Wu, P.X. Shen, J. He, et al., Science. 355 (2017) 499–503. doi: 10.1126/science.aal5175
J.L. Hofstra, A.H. Cherney, C.M. Ordner, S.E. Reisman, J. Am. Chem. Soc. 140 (2018) 139–142. doi: 10.1021/jacs.7b11707
J.F. Bai, K. Yasumoto, T. Kano, K. Maruoka, Angew. Chem. Int. Ed. 58 (2019) 8898–8901. doi: 10.1002/anie.201904520
T. Hong, Z. Zhang, Y. Sun, et al., J. Am. Chem. Soc. 142 (2020) 10244–10249. doi: 10.1021/jacs.0c01563
L. Jin, Q.J. Yao, P.P. Xie, et al., Chem 6 (2020) 497–511. doi: 10.1016/j.chempr.2019.12.011
J. Liu, H. Gong, S. Zhu, Angew. Chem. Int. Ed. 60 (2021) 4060–4064. doi: 10.1002/anie.202012614
P. Xiong, M. Hemming, S.I. Ivlev, E. Meggers, J. Am. Chem. Soc. 144 (2022) 6964–6971. doi: 10.1021/jacs.2c01686
X. Li, M. Yuan, F. Chen, et al., Chem 9 (2023) 154–169. doi: 10.1016/j.chempr.2022.09.020
R.F. Heck, J. Am. Chem. Soc. 90 (1968) 5518–5526. doi: 10.1021/ja01022a034
N.E. Carpenter, D.J. Kucera, L.E. Overman, J. Org. Chem. 54 (1989) 5846–5848. doi: 10.1021/jo00286a009
Y. Sato, M. Sodeoka, M. Shibasaki, J. Org. Chem. 54 (1989) 4738–4739. doi: 10.1021/jo00281a007
M. Shibasaki, E.M. Vogl, T. Ohshima, Adv. Synth. Catal. 346 (2004) 1533–1552. doi: 10.1002/adsc.200404203
H. Li, C. Ding, B. Xu, X. Hou, Acta Chim. Sin. 72 (2014) 765–770. doi: 10.6023/A14040329
B. Xiang, T.F. Xu, L. Wu, et al., J. Org. Chem. 81 (2016) 3929–3935. doi: 10.1021/acs.joc.6b00358
L. Jin, P. Zhang, Y. Li, X. Yu, B.F. Shi, J. Am. Chem. Soc. 143 (2021) 12335–12344. doi: 10.1021/jacs.1c06236
S. Li, Q. Chen, Z.M. Zhang, J. Zhang, Green Synth. Catal. 2 (2021) 374–376. doi: 10.1016/j.gresc.2021.10.006
K. Liang, Q. Zhang, C. Guo, Sci. Adv. 8 (2022) eadd7134. doi: 10.1126/sciadv.add7134
R.R. Liu, D.J. Wang, L. Wu, et al., ACS Catal. 5 (2015) 6524–6528. doi: 10.1021/acscatal.5b01793
L.J. Xiao, C.Y. Zhao, L. Cheng, et al., Angew. Chem. Int. Ed. 57 (2018) 3396–3400. doi: 10.1002/anie.201713333
T. Hashimoto, H. Kimura, K. Maruoka, Angew. Chem. Int. Ed. 49 (2010) 6844–6847. doi: 10.1002/anie.201003600
L. Cui, L. Zhang, S. Luo, J.P. Cheng, Eur. J. Org. Chem. 2014 (2014) 3540–3545. doi: 10.1002/ejoc.201402353
H.H. Zhang, Y.M. Wang, Y.W. Xie, et al., J. Org. Chem. 79 (2014) 7141–7151. doi: 10.1021/jo501293m
J.H. Xue, M. Shi, F. Yu, et al., Org. Lett. 18 (2016) 3874–3877. doi: 10.1021/acs.orglett.6b01880
X.Y. Li, W.T. Hu, Q.J. Xiong, et al., Adv. Synth. Catal. 361 (2019) 1803–1807. doi: 10.1002/adsc.201801705
K.S. Brown, C. Djerassi, J. Am. Chem. Soc. 86 (1964) 2451–2463. doi: 10.1021/ja01066a031
S. Atarashi, S. Yokohama, K.I. Yamazaki, et al., Chem. Pharm. Bull. 35 (1987) 1896–1902. doi: 10.1248/cpb.35.1896
S.D. McAllister, G. Rizvi, S. Anavi-Goffer, et al., J. Med. Chem. 46 (2003) 5139–5152. doi: 10.1021/jm0302647
S.S. Michael, Patent, WO 2012/151440 A1, 2012.
R.I. Storer, D.E. Carrera, Y. Ni, D.W.C. MacMillan, J. Am. Chem. Soc. 128 (2006) 84–86. doi: 10.1021/ja057222n
X. Zhang, B. Xu, M.H. Xu, Org. Chem. Front. 3 (2016) 944–948. doi: 10.1039/C6QO00191B
H.C. Shen, Y.F. Wu, Y. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 2372–2376. doi: 10.1002/anie.201712350
M. Viji, J. Sim, S. Li, et al., Adv. Synth. Catal. 360 (2018) 4464–4469. doi: 10.1002/adsc.201800870
Y.H. Geng, Y.Z. Hua, S.K. Jia, M.C. Wang, Chem. Eur. J. 27 (2021) 5130–5135. doi: 10.1002/chem.202100284
J. Zhang, J. Wei, W.Y. Ding, et al., J. Am. Chem. Soc. 143 (2021) 6382–6387. doi: 10.1021/jacs.1c02808
M. Hatano, Y. Goto, A. Izumiseki, M. Akakura, K. Ishihara, J. Am. Chem. Soc. 137 (2015) 13472–13475. doi: 10.1021/jacs.5b08693
Q.X. Zhang, Y. Li, J. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 4550–4556. doi: 10.1002/anie.201915226
Q. Zhao, Y. Li, Q.X. Zhang, J.P. Cheng, X. Li, Angew. Chem. Int. Ed. 60 (2021) 17608–17614. doi: 10.1002/anie.202104788
T. Akiyama, Chem. Rev. 107 (2007) 5744–5758. doi: 10.1021/cr068374j
M. Terada, Synthesis (Mass). 2010 (2010) 1929–1982. doi: 10.1055/s-0029-1218801
T. Akiyama, K. Mori, Chem. Rev. 115 (2015) 9277–9306. doi: 10.1021/acs.chemrev.5b00041
T. James, M. van Gemmeren, B. List, Chem. Rev. 115 (2015) 9388–9409. doi: 10.1021/acs.chemrev.5b00128
M.J. Frisch, G.W. Trucks, H.B. Schegel, et al., Gaussian16, Revision C.01, 2016.
F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 7 (2005) 3297–3305. doi: 10.1039/b508541a
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 132 (2010) 154104. doi: 10.1063/1.3382344
Y. Zhao, D.G. Truhlar, Acc. Chem. Res. 41 (2008) 157–167. doi: 10.1021/ar700111a
Y. Zhao, D.G. Truhlar, Chem. Phys. Lett. 502 (2011) 1–13. doi: 10.1007/978-3-642-24559-6_1
F.L. Hirshfeld, Theoret. Chim. Acta 44 (1977) 129–138. doi: 10.1007/BF00549096
S. Emamian, T. Lu, H. Kruse, H. Emamian, J. Comput. Chem. 40 (2019) 2868–2881. doi: 10.1002/jcc.26068

Scheme 2 Scope of the benzoxazinones. The reactions were carried out with 1 (0.1 mmol), 2 (0.12 mmol), B(C6F5)3 (2 mol%), (S)-4a (2 mol%) and 5 Å MS (50.0 mg) in DCM (0.5 mL) at −20 ℃ under argon for 48 h, yields of isolated products. The e.r. values were determined by HLPC analysis. aAt r.t. bAt −60 ℃. c(S)-5b (2 mol%) was used.

Scheme 3 Scope of the alkenes. The reactions were carried out with 1 (0.1 mmol), 2 (0.12 mmol), B(C6F5)3 (2 mol%), (S)-4a (2 mol%) and 5 Å MS (50.0 mg) in DCM (0.5 mL) at −20 ℃ under argon for 48 h, yields of isolated products. The e.r. values were determined by HLPC analysis. aZ/E ratio, determined by using 1H NMR.

Scheme 6 (a) Calculated free energy surface for Borane/chiral Brønsted acid-catalyzed asymmetric conjugate addition of addition of benzoxazinone with diphenylethylene. (b) Calculated structures and the relative Gibbs free energy for the lowest-lying transition states.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

