
Figure 1.
SEM images of (a) pure C-Mn3O4 and (c) 1NOC-Mn3O4; TEM images of (b) pure C-Mn3O4 and (d) 1NOC-Mn3O4; (e) EDS elemental mapping images C, N, O and Mn in the 1NOC-Mn3O4; HRTEM image of (f) 1NOC-Mn3O4.
Covalent organic frameworks doped with manganese-metal organic framework for peroxymonosulfate activation
Yinyin Xu , Yuanyuan Li , Jingbo Feng , Chen Wang , Yan Zhang , Yukun Wang , Xiuwen Cheng

The rapid economic and industrial development of modern society have produced and used a large of toxic chemicals, such as organic dyes, endocrine disruptors, resulting in serious water pollution [1]. Since the serious threat to ecosystems of these organic pollutants [2,3], there is an urgent need to develop technologies to effectively remove them. In recent years, advanced oxidation processes (AOPs) based on persulfate could convert organic pollutants into less harmful intermediates, which was considered an effective method to degrade organic pollutants. Compared with thermal, radiation, ultrasonic and homogeneous catalysts for persulfate activation, transition metal and carbon heterogeneous catalysts were widely used for their low energy consumption, low ion leaching and high oxidation efficiency [4,5]. Therefore, the design of effective heterogeneous catalysts was the key to the catalytic degradation of organic pollutants.
Manganese oxide (MnOx) was widely used in peroxymonosulfate (PMS) activation due to its low environmental toxicity, high natural abundance and various oxidation states [6]. However, the stability and conductivity of a single MnOx were poor [7]. To solve this problem, MnOx@carbon materials with the manganese-organic framework (Mn-MOF) as its precursor stood out. The Mn-MOF was a kind of porous material with unique physical and chemical properties and adjustable structure [8]. The derived MnOx@carbon hybrid inherited the characteristics of MOF itself, effectively restricts crystal growth and had abundant defects, which was conducive to the activation of PMS [9]. Zhao et al. prepared MnOx@C nanosheets by in-situ carbonization of Mn-MOF, which effectively activated PMS to degrade 4-aminobenzoic acid ethyl ester [10].
Moreover, heteroatoms (N, O, S, etc.) doping could be achieved by directly carbonizing the precursor containing heteroatoms [11]. The heteroatom doping could break the inertia of carbon catalysts, engender unique active sites, improve the efficiency of electron transfer, and further enhance the PMS activation [12]. The ability of Mn-MOF-derived hybrids to activate PMS to degrade target organic pollutants could be improved by heteroatom doping. Xie et al. added melamine as an additional nitrogen source and Mn-MOF as a precursor to prepare N-doped Mn2O3@porous carbon to activate PMS, showing excellent catalytic activity [8]. However, little attention seemed to have been paid to covalent organic frameworks (COFs) as precursors for heteroatom doping. COFs are a kind of multi-space nanomaterial composed of light elements with definite structures [13]. In our previous paper, COFs remained morphologically unchanged during carbonization, providing more active sites (graphite N and ketone groups) and improving electron transfer efficiency, which significantly enhanced PMS activation [14]. Therefore, adding COFs as the precursor of heteroatom doping was considered a method to improve the catalytic performance. Herein, the nitrogen and oxygen co-doped Mn3O4@porous carbon material (NOC-Mn3O4) was prepared by hydrothermal treatment and co-carbonization of Mn-MOF and COF, and was used for the degradation of target organic pollutants bisphenol A (BPA) and rhodamine B (RhB).
Details of chemicals and characterizations were available in Texts S1-S2 and Table S1 (Supporting information). The NOC-Mn3O4 was synthesized with a solvothermal method and the subsequent carbonization was based on the previous report (the detailed synthesis process could be found in Text S3 in Supporting information) [10]. The 1,3,5-tris(4-aminophenyl)benzene and 2,5-dimethoxyterephaldehyde were used as monomers to synthesize COF. The samples obtained after carbonizing Mn-MOF with different amounts of COF were named C-Mn3O4, 0.5NOC-Mn3O4, 1NOC-Mn3O4 and 2NOC-Mn3O4.
As shown in Fig. S1 (Supporting information), Mn-MOF presented a clear layered structure. The appearance of spherical aggregates after complex COF indicated the successful combination of Mn-MOF and COF. After carbonization, the C-Mn3O4 derived from Mn-MOF maintained a layered structure and was composed of a large number of nanoparticles (Figs. 1a and b). As seen in Figs. 1c and d, 1NOC-Mn3O4 effectively prevented the agglomeration of nanoparticles and the doping of COF improved the porosity. Energy-dispersive X-ray spectroscopy (EDS) mappings suggested that C, N, O and Mn were distributed in 1NOC-Mn3O4 (Fig. 1e). Table S2 (Supporting information) compared Brunauer-Emmett-Teller (BET) specific surface area and pore size distribution of C-Mn3O4 and 1NOC-Mn3O4, both of which were mesoporous structures. In addition, the BET surface area of 1NOC-Mn3O4 (31.946 m2/g) was larger than that of C-Mn3O4 (14.009 m2/g), which was consistent with scanning electron microscopy (SEM). The detailed discussion could be found in Text S4 and Fig. S2 (Supporting information). As shown in Fig. 1f, the appearance of lattice fringe (0.248 nm, 0.313 nm, 0.276 nm and 0.492 nm) in 1NOC-Mn3O4 indicated the existence of Mn3O4 nanoparticles, which was consistent with X-ray diffraction (XRD) [15]. Besides, the thermal gravity results showed that 1NOC-Mn3O4 had good thermal stability (detailed discussion could be found in Text S5 and Fig. S3 in Supporting information).
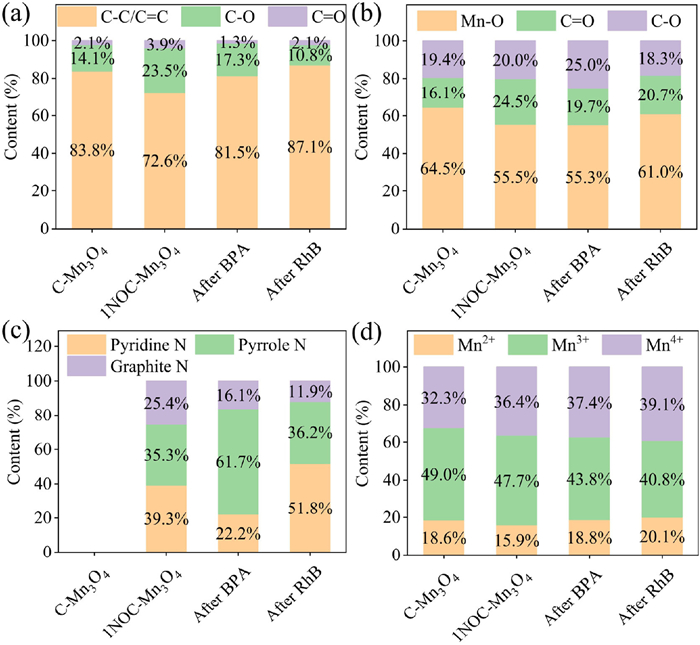
As shown in Fig. S4 (Supporting information), the XRD patterns of C-Mn3O4 and NOC-Mn3O4 samples corresponded to Mn3O4 (PDF #80-0382). Compared with the XRD spectra of C-Mn3O4, a slight shift of the characteristic peak was observed in 1NOC-Mn3O4, which was attributed to COF doping. The detailed discussion could be found in Text S6 (Supporting information). The structures of catalysts were further studied by Fourier transform infrared spectroscopy (FT-IR). As shown in Fig. S5 (Supporting information), the characteristic peaks of Mn-O and C=O were observed in 1NOC-Mn3O4. It was worth noting that C=O in 1NOC-Mn3O4 was partly derived from carbonized COF and partly from C-Mn3O4. The detailed discussion could be found in Text S7 (Supporting information). In addition, the elemental composition of C-Mn3O4 and 1NOC-Mn3O4 was analyzed by X-ray photoelectron spectroscopy (XPS) (Figs. 2a-d and Fig. S6 in Supporting information). The C 1s, O 1s and Mn 2p appeared in the full XPS spectrum of C-Mn3O4 and 1NOC-Mn3O4. Due to the doping of COF, 1NOC-Mn3O4 had a peak of N 1s. In the C 1s spectrum of 1NOC-Mn3O4, the peaks at 284.8 eV, 285.8 eV and 288.4 eV corresponded to C-C/C=C, C-O and C=O. The O 1s of 1NOC-Mn3O4 could be deconvoluted into Mn-O (530.0 eV), C=O (531.2 eV) and C-O (532.7 eV) [16]. The C=O content of 1NOC-Mn3O4 (3.9% in C 1s, 24.5% in O 1s) was higher than that of C-Mn3O4 (2.1% in C 1s, 16.1% in O 1s), indicating that the doping of COF was conducive to the formation of active group C=O, which was consistent with the FT-IR result. Meanwhile, the N 1s spectrum of 1NOC-Mn3O4 showed three peaks at 399.5 eV, 400.2 eV and 401.2 eV were ascribed to pyridine N, pyrrole N and graphite N. However, no N 1s was found in C-Mn3O4, which proved that the N element in 1NOC-Mn3O4 came from COF doping. The Mn 2p spectra of 1NOC-Mn3O4 were fitted into three components corresponding to Mn2+ (641.0 eV, 651.7 eV), Mn3+ (642.4 eV, 653.7 eV) and Mn4+ (643.9 eV, 654.6 eV) [17]. In summary, the N and O doping in COF might be the reason for improving the catalytic ability.
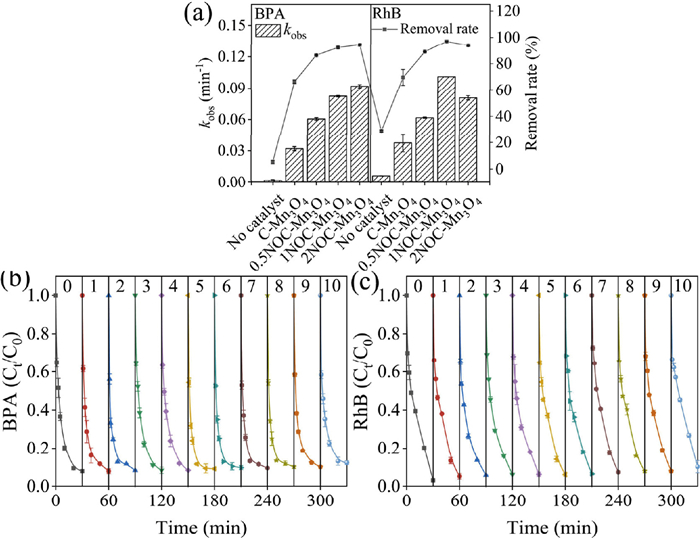
Before the catalytic performance of the catalysts could be evaluated, it needed to be determined whether the BPA and RhB could be removal by adsorption of the catalysts. Fig. S7 (Supporting information) showed the removal rates of BPA and RhB under different conditions (i.e., COF adsorption, COF/PMS system, NOC adsorption, NOC/PMS system, 0.5NOC-Mn3O4 adsorption, 1NOC-Mn3O4 adsorption, 2NOC-Mn3O4 adsorption,). The results showed that the composite catalysts had limited adsorption performance (less than 16%) for BPA and RhB. The detailed discussion could be found in Text S8 (Supporting information). The catalytic performance of different catalysts for PMS activation was evaluated by BPA and RhB degradation experiment (Fig. 3a). The original data was shown in Fig. S8 (Supporting information). The detailed degradation experiment process could be found in Text S9 (Supporting information). For BPA, the removal rates within 30 min were 5.4%, and 66.6% by PMS alone and C-Mn3O4/PMS system, respectively. The corresponding pseudo-first-order reaction rate constant (kobs) were 0.002 min−1 and 0.032 min−1. Similarly, the removal rates of RhB were 28.9% and 69.8% by PMS alone and C-Mn3O4/PMS system, respectively. The corresponding kobs within 30 min were 0.006 min−1 and 0.037 min−1. These results indicated that PMS alone and C-Mn3O4/PMS system without COF doping had limited PMS activation ability. When doped with COF, the degradation of BPA was improved with removal rates of 86.4% by 0.5NOC-Mn3O4/PMS system, 92.1% by 1NOC-Mn3O4/PMS system and 94.2% by 2NOC-Mn3O4/PMS system, which were 1.3, 1.4 and 1.4 times higher than those of C-Mn3O4/PMS system. The corresponding kobs were 0.060 min−1, 0.082 min−1 and 0.092 min−1, which were 1.9, 2.6 and 2.9 times higher than those of C-Mn3O4/PMS system. Similarly, the improved degradation of RhB was achieved with removal rates of 88.9% by 0.5NOC-Mn3O4/PMS system, 96.9% by 1NOC-Mn3O4/PMS system and 93.9% by 2NOC-Mn3O4/PMS system, which were 1.3, 1.4 and 1.3 times higher than those of C-Mn3O4/PMS system. The corresponding kobs were 0.062 min−1, 0.101 min−1 and 0.081 min−1, which were 1.7, 2.7 and 2.2 times higher than those of C-Mn3O4/PMS system. These results suggested that COF-doped C-Mn3O4 could activate PMS more efficiently and then remove BPA and RhB in water. In addition, with the increase of COF content, the removal rate of BPA and RhB degradation by 1NOC-Mn3O4/PMS system was higher than that by 0.5NOC-Mn3O4/PMS system. This proved that the increase of COF content in the precursor material could significantly improve the catalytic activity. In our previous study, COF doping could provide more active sites (graphite N and ketone groups) and accelerate electron transfer, and then significantly improve BPA and RhB degradation. However, with the content of COF increasing, the removal rate of BPA was not obviously increased, and the removal rate of RhB slightly decreased. This might be attributed to the excessive introduction of COF which might encase the active sites and thus lead to the reduction in catalytic activity [18]. From an economic point of view, the addition of the expensive COF did not provide a significant improvement, so 1NOC-Mn3O4 was selected for the subsequent research.
To further investigate the catalytic performance of 1NOC-Mn3O4/PMS system, the effects of several operating parameters on BPA and RhB degradation were examined (Figs. S9-S11 in Supporting information), including catalyst dosage, PMS dosage, BPA and RhB concentration, reaction temperature and initial pH. In short, the optimized systems with 0.20 g/L 1NOC-Mn3O4 and 0.20 g/L PMS for removing 20 mg/L BPA at unadjusted pH (6.35), 0.20 g/L 1NOC-Mn3O4 and 0.30 g/L PMS for removing 10 mg/L RhB at unadjusted pH (5.30) were recommended. The activation energy (Ea) of 1NOC-Mn3O4 for PMS activation during BPA and RhB degradation could be calculated to be 2.26 kJ/mol and 18.7 kJ/mol. More than 90% BPA and RhB could be removed in the wide range of pH 3-10. The detailed discussion could be found in Text S10 (Supporting information). In addition, the 1NOC-Mn3O4/PMS system could effectively remove tetracycline, congo red, malachite green and crystalline violet in water, indicating that the general applicability of 1NOC-Mn3O4/PMS system to organic pollutants. The detailed discussion could be found in Text S11 and Fig. S12 (Supporting information).
The actual water generally contained a variety of ions and natural organic matter (NOM). Therefore, the effects of NOM representing humic acid (HA) and different ions on BPA and RhB degradation were investigated to assess the adaptability of the 1NOC-Mn3O4/PMS system (Figs. S13-15 in Supporting information). Briefly, CO32− and phosphate (H2PO4−, HPO42− and PO43−) exerted a strong inhibitory effect on the catalytic performance of the 1NOC-Mn3O4/PMS system, while other ions, including Cl−, NO3−, SO42−, Ca2+ and Mg2+, had an acceptable effect on BPA and RhB degradation. This strong inhibitory effect of CO32− was attributed to the fact that CO32− was a potent quencher of SO4•‒ and •OH [19]. Furthermore, CO32− reacted with Mn2+ in solution to form MnCO3 precipitation, which was to disadvantage of pollutants degradation [20]. The inhibitory effect of phosphate was attributed to the fact that phosphate not only could react with ROS but also could act as a chelator attached to the 1NOC-Mn3O4 surface, thereby reducing the catalytic activity [21]. Besides, both HCO3− and HA inhibited the degradation of BPA and RhB, but the degree was different. The inhibitory effect of HCO3− on the degradation of RhB was stronger than that of BPA, and the inhibitory effect of HA on the degradation of BPA was greater than that of RhB. The inhibitory effect of BPA by HA was attributed to the adsorption of HA onto the 1NOC-Mn3O4 surface, preventing the reaction between the PMS and the active sites. However, RhB degradation was slightly affected. The phenomenon was attributed to the quinone functional groups could effectively activate PMS to degrade RhB [22]. In addition, BPA and RhB were effectively degraded in actual water, including Tap water, Lake water and Yellow River water. The detailed discussion could be found in Text S12 (Supporting information).
Ten reuse experiments were conducted to evaluate the reusability of 1NOC-Mn3O4/PMS system. After each reuse experiment, the removal rates of BPA and RhB were shown in Figs. 3b and c. It was clear that the removal rates of BPA and RhB decreased after ten reuse experiments but still maintained over 87%, indicating that 1NOC-Mn3O4 was an efficient and durable PMS catalyst. This slight decrease was associated with the occupation and loss of active sites. The ICP-OES test was performed after each reuse experiment, and the results were shown in Fig. S16a (Supporting information). The maximum leaching concentration for Mn was 21.8 mg/L and 17.7 mg/L after BPA and RhB degradation. This might be due to the repeated acid etching of 1NOC-Mn3O4 in the presence of PMS, as well as the transformation of Mn between different valence states, resulting in the loss of Mn [23]. In order to distinguish between homogeneous and heterogeneous reactions, solutions with the highest Mn2+ concentrations were configured to simulate the homogeneous catalytic reactions which could be caused by the leaching of Mn2+. As shown in Fig. S16b (Supporting information), the removal rate of BPA and RhB by Mn2+/PMS system was 24.3% and 28.6%, respectively. Meanwhile, the removal rate of BPA and RhB by PMS was 5.4% and 28.9%, respectively (Fig. 3a). The results showed that only 18.9% of BPA was removed in the Mn2+/PMS system, without significant effect of RhB (0.3% as an error), indicating that the heterogeneous reaction played a major role in the 1NOC-Mn3O4/PMS system. In addition, SEM images of fresh and used 1NOC-Mn3O4 were shown in Figs. S16c-e (Supporting information). Compared to fresh 1NOC-Mn3O4, there was some damage to the morphology of the used 1NOC-Mn3O4. The above results showed that 1NOC-Mn3O4 obtained a certain stability.
The tert-butanol (TBA), methanol (MeOH), furfuryl alcohol (FFA) and p-benzoquinone (p-BQ) were used as scavengers to study the reactive oxygen species (ROS: •OH, SO4•‒, 1O2 and O2•−) produced in 1NOC-Mn3O4/PMS system (Fig. 4a). When the concentrations of TBA, MeOH, FFA and p-BQ were 2.00 mol/L/2.00 mol/L, 2.00 mol/L/2.00 mol/L, 4.0 mmol/L/10 mmol/L and 4.0 mmol/L/10 mmol/L, the removal rates of BPA/RhB were reduced by 10.8%/30.3%, 11.7%/7.8%, 44.5%/13.3% and 65.6%/10.9%. This suggested that 1O2 and O2•− played an important role in the degradation of BPA by 1NOC-Mn3O4/PMS, while •OH played an important role in RhB degradation (The effect of other concentrations of scavengers on the degradation of BPA and RhB were shown in Text S13 and Fig. S17 (in Supporting information). The electron paramagnetic resonance (EPR) test further proved the presence of ROS. As shown in Fig. 4b, when the NOC-Mn3O4/PMS system reacted for 1 min, there were clear characteristic signals of the DMPO-•OH, DMPO-SO4•‒, DMPO-O2•− and TEMP-1O2, and the signal intensity increased at 10 min, indicating •OH, SO4•‒, O2•− and 1O2 existed in the NOC-Mn3O4/PMS system and produced continuously.

The electrochemical impedance spectroscopy (EIS) showed that 1NOC-Mn3O4 had a smaller Nyquist plot diameter than C-Mn3O4, proving that 1NOC-Mn3O4 had a better electrical conductivity (the inset Fig. 4c). As illustrated in the linear sweep voltammetry curves (LSV) (Fig. 4d), the current of the 1NOC-Mn3O4 electrode was greater than C-Mn3O4, showing a stronger redox ability. The results indicated that COF doping could significantly promote electron transfer ability. With the addition of PMS, the current increased significantly, indicating that more electron transfer occurred in the process of PMS activation. Then, when BPA/RhB was added, the current response was further enhanced, indicating the transfer of electrons from BPA/RhB to the surface complex formed by the PMS and 1NOC-Mn3O4. Meanwhile, the electron transfer process of pollutant, PMS and 1NOC-Mn3O4 was studied by the I-t curves (Fig. 4c). Significant current changes were observed when PMS and BPA/RhB were added, respectively. This indicated that the electron transfer process occurred among the PMS, 1NOC-Mn3O4 and pollutants, resulting in the degradation of the BPA and RhB. Therefore, electron transfer occurred in the 1NOC-Mn3O4/PMS system, and COF doping accelerated electron transfer.
To further investigate the active sites in the 1NOC-Mn3O4/PMS system, XPS tests were performed on 1NOC-Mn3O4 after the degradation reaction of BPA and RhB (Figs. 2a-d). It was found that the C=O content in C 1s decreased from 3.9% to 1.3%/2.1% after BPA/RhB degradation reaction. Similarly, the C=O content in O 1s decreased from 24.5% to 19.7%/20.7%. Therefore, the C=O was the active site for 1NOC-Mn3O4 to activate PMS. Meanwhile, the content of graphite N decreased from 25.4% to 16.1%/11.9% after BPA/RhB reaction, suggesting graphite N was involved in the reaction. Furthermore, the Mn3+ content decreased from 47.7% to 43.8%/40.8% after the degradation reaction of BPA/RhB, while the Mn4+ content increased from 36.4% to 37.4%/39.1%. This phenomenon was due to the flow of electrons from Mn3+ on the 1NOC-Mn3O4 surface to PMS to produce ROS to degrade target pollutants [24]. Moreover, the Mn2+ content increased from 15.9% to 18.8%/20.1% after BPA/RhB degradation reaction. The results indicated that the Mn2+/Mn3+/Mn4+ cycle was also involved in PMS activation.
Based on the above analysis, the possible mechanism of degradation of BPA and RhB by the 1NOC-Mn3O4-activated PMS was proposed. Generally, the 1NOC-Mn3O4/PMS system degraded BPA and RhB through ROS reactions and electron transfer. Firstly, Mn2+/Mn3+ activated PMS to produce Mn3+/Mn4+ and SO4•‒/•OH. PMS should further reduce Mn3+/Mn4+ to Mn2+/Mn3+ and generate SO5•−, thus forming the Mn2+/Mn3+/Mn4+ cycle (Eqs. 1-3) [10]. Next, C=O induced the decomposition of PMS by providing electrons to PMS adsorbed on the 1NOC-Mn3O4 surface to produce SO5•−, which was readily transformed to produce 1O2 (Eqs. 4 and 5) [25]. Graphite N changed the electron density of the adjacent carbon, enhancing the adsorption between PMS and 1NOC-Mn3O4, which in turn induced the production of 1O2 (Eqs. 4 and 5) [26]. O2•− was present as an intermediate in the production of 1O2. Finally, graphite N acquired electrons from the adjacent carbon, and the PMS adsorbed on the 1NOC-Mn3O4 surface accepted the electrons to produce SO4•‒ and •OH (Eqs. 6 and 7) [27]. Therefore, the •OH, SO4•‒, O2•−, 1O2 and electron transfer processes in the 1NOC-Mn3O4/PMS system together led to the degradation of BPA and RhB.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
|
|
(6) |
|
|
(7) |
The mineralization degree of BPA and RhB in the 1NOC-Mn3O4/PMS system was investigated. The TOC removal of BPA and RhB by the 1NOC-Mn3O4/PMS system was 51.5% and 61.8%, respectively, indicating that the 1NOC-Mn3O4/PMS system achieved effective mineralization of BPA and RhB. Liquid chromatography-mass spectrometry (LC-MS) was used to detect the degradation intermediates of BPA and RhB. The detailed information of detected intermediates as shown in Figs. S18, S19 and Tables S3, S4 (Supporting information). A possible BPA degradation pathway was proposed based on the previous literature (Fig. S20 in Supporting information) [28,29]. BPA (B1, m/z = 228) could break the C-C bond to form B2/B3 (m/z = 216/214), further open the benzene ring to form B5-B7 (m/z = 108, 94, 92), and could dehydroxyl to form B4 (m/z = 196). These intermediates could open rings to form B8-B22 (m/z = 128, 118, 114, 98, 90, 88, 86, 60), and further mineralize to form harmless products finally. According to the previous literature, the degradation pathway of RhB in the 1NOC-Mn3O4/PMS system was proposed (Fig. S21 in Supporting information) [30–35]. Generally, on the one hand, RhB (R1, m/z = 443) formed R2 (m/z = 459) through methyl oxidation, on the other hand, the R3/R4 (m/z = 415), R5/R6/R7 (m/z = 387), R8/R9 (m/z = 359) and R10 (m/z = 331) were formed through successive N-de-ethylation processes. Then, intermediates with smaller molecular weights R11-R26 (m/z = 317, 318, 301, 298, 278, 274, 269, 258, 255, 243, 230, 213, 212, 166, 122, 109) were formed through a series of reactions. Further, these intermediates formed some ring-opening products through ring-opening reactions. Finally, these products were destroyed to form inorganic substances. In addition, Toxicity Estimation Software Tool (T.E.S.T.) was used to evaluate BPA, RhB and their degradation intermediates (Fig. S22 and Text S14 in Supporting information). The results showed that the 1NOC-Mn3O4/PMS system could reduce the comprehensive toxicity of BPA and RhB.
In conclusion, the prepared 1NOC-Mn3O4 showed excellent catalytic performance in activating PMS to degrade BPA and RhB. Meanwhile, the 1NOC-Mn3O4/PMS system degraded target pollutants in radical and non-radical ways. More importantly, the COF doping was good for the formation of C=O and graphite N, the increase of the BET surface area, and the acceleration of electron transfer.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors are grateful for the Fundamental Research Funds for the Central Universities (No. lzujbky-2022-59), National Natural Science Foundation of China (No. 51978319).
Supplementary material associated with this article can be found, in the online version, at doi:
X. Yang, J. Duan, X. Zhang, et al., Chin. Chem. Lett. 33 (2022) 3792–3796. doi: 10.1016/j.cclet.2021.11.031
Y. Zhou, J. He, J. Lu, et al., Chin. Chem. Lett. 31 (2020) 2623–2626. doi: 10.1016/j.cclet.2020.02.008
X. Shi, P. Hong, H. Huang, et al., J. Colloid Interface Sci. 610 (2022) 751–765. doi: 10.1016/j.jcis.2021.11.118
X. Zhou, Q. Zhao, J. Wang, et al., J. Hazard. Mater. 434 (2022) 128773. doi: 10.1016/j.jhazmat.2022.128773
Y. Zhao, X. Zhan, Y. Sun, et al., Chemosphere 310 (2023) 136937. doi: 10.1016/j.chemosphere.2022.136937
S. Ghanbari, A. Fatehizadeh, E. Taheri, et al., Environ. Res. 215 (2022) 114316. doi: 10.1016/j.envres.2022.114316
K. Zhang, H. Zeng, J. Feng, et al., Food Chem. Toxicol. 162 (2022) 112908. doi: 10.1016/j.fct.2022.112908
Z. Xie, Z. Lyu, J. Wang, et al., Chem. Eng. J. 429 (2022) 132299. doi: 10.1016/j.cej.2021.132299
K. Sun, J. Pang, Y. Zheng, et al., J. Alloy. Compd. 923 (2022) 166470. doi: 10.1016/j.jallcom.2022.166470
Y. Zhao, X. Zhan, H. Wang, et al., Chem. Eng. J. 433 (2022) 133806. doi: 10.1016/j.cej.2021.133806
S. Liu, Z. Zhang, F. Huang, et al., Appl. Catal. B 286 (2021) 119921. doi: 10.1016/j.apcatb.2021.119921
Z.Y. Choong, K.A. Lin, G. Lisak, et al., J. Hazard. Mater. 426 (2022) 128077. doi: 10.1016/j.jhazmat.2021.128077
M. Cao, J. Lei, J. Zhang, et al., J. Clean. Prod. 375 (2022) 134114. doi: 10.1016/j.jclepro.2022.134114
Y. Li, J. Feng, Y. Zhang, et al., Chemosphere 311 (2023) 137038. doi: 10.1016/j.chemosphere.2022.137038
J. Chen, B. Bai, J. Lei, et al., Chem. Eng. Sci. 263 (2022) 118065. doi: 10.1016/j.ces.2022.118065
C.M. Hung, C.W. Chen, C.P. Huang, et al., Environ. Pollut. 311 (2022) 119984. doi: 10.1016/j.envpol.2022.119984
Q. Deng, X. Zhang, L. Chang, et al., Sep. Purif. Technol. 294 (2022) 121182. doi: 10.1016/j.seppur.2022.121182
L. Zou, X. Zhu, L. Lu, et al., J. Hazard Mater. 419 (2021) 126427. doi: 10.1016/j.jhazmat.2021.126427
B. Gao, S. Zhu, J. Gu, et al., J. Hazard Mater. 431 (2022) 128549. doi: 10.1016/j.jhazmat.2022.128549
J. Fan, Q. Wang, W. Yan, et al., J. Hazard Mater. 434 (2022) 128818. doi: 10.1016/j.jhazmat.2022.128818
W. Zhao, Z. Duan, Z. Zheng, et al., Chem. Eng. J. 433 (2022) 133742. doi: 10.1016/j.cej.2021.133742
G. Fang, J. Gao, D.D. Dionysiou, et al., Environ Sci. Technol. 47 (2013) 4605–4611. doi: 10.1021/es400262n
J. Liang, L. Fu, K. Gao, et al., Appl. Catal. B 315 (2022) 121542. doi: 10.1016/j.apcatb.2022.121542
K. Li, S. Xu, X. Liu, et al., Chem. Eng. J. 438 (2022) 135630. doi: 10.1016/j.cej.2022.135630
Q. Wang, Y. Jiang, S. Yang, et al., Chem. Eng. J. 445 (2022) 136806. doi: 10.1016/j.cej.2022.136806
H. Zhu, A. Guo, S. Wang, et al., Chem. Eng. J. 450 (2022) 138428. doi: 10.1016/j.cej.2022.138428
N. Li, J. Ye, H. Dai, et al., Water Res. 235 (2023) 119926. doi: 10.1016/j.watres.2023.119926
C. Yin, Q. Xia, J. Zhou, et al., Sep. Purif. Technol. 280 (2022) 119924. doi: 10.1016/j.seppur.2021.119924
J. Dan, Q. Wang, P. Rao, et al., Chem. Eng. J. 429 (2022) 132189. doi: 10.1016/j.cej.2021.132189
W. Li, Y. Zhang, P. Zhao, et al., J. Hazard. Mater. 393 (2020) 122399. doi: 10.1016/j.jhazmat.2020.122399
D. Liu, D. Chen, Z. Hao, et al., Chemosphere 307 (2022) 135935. doi: 10.1016/j.chemosphere.2022.135935
J. Tan, C. Xu, X. Zhang, et al., Sep. Purif. Technol. 289 (2022) 120812. doi: 10.1016/j.seppur.2022.120812
X. Tong, W. Jia, Y. Li, et al., J. Taiwan Inst. Chem. Eng. 102 (2019) 92–98. doi: 10.1016/j.jtice.2019.05.008
Y. Xu, E. Hu, D. Xu, et al., Sep. Purif. Technol. 274 (2021) 119081. doi: 10.1016/j.seppur.2021.119081
Q. Zhong, J. Liu, J. Wang, et al., Chemosphere 300 (2022) 134564. doi: 10.1016/j.chemosphere.2022.134564

Figure 1 SEM images of (a) pure C-Mn3O4 and (c) 1NOC-Mn3O4; TEM images of (b) pure C-Mn3O4 and (d) 1NOC-Mn3O4; (e) EDS elemental mapping images C, N, O and Mn in the 1NOC-Mn3O4; HRTEM image of (f) 1NOC-Mn3O4.

Figure 2 The XPS configurations of (a) C 1s, (b) O 1s, (c) N 1s, (d) Mn 2p of C-Mn3O4, 1NOC-Mn3O4 and 1NOC-Mn3O4 after degradation reaction.

Figure 3 (a) The catalytic performance of different catalysts/PMS system; Reusability of 1NOC-Mn3O4 for the degradation of (b) BPA and (c) RhB. Reaction condition: [catalyst] = 0.20 g/L, [PMS] = 0.20 g/L (BPA degradation), [PMS] = 0.30 g/L (RhB degradation), [BPA] = 20 mg/L, [RhB] = 10 mg/L, unadjusted pH (BPA: 6.35, RhB: 5.30), Temperature = 25 ± 3 ℃.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: