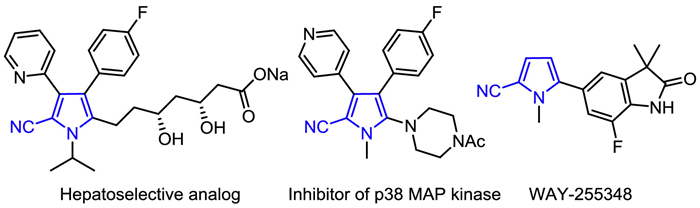
Figure 1.
Bioactive molecules with 2-cyanopyrrole moiety.
Oxidative cyclization of allyl compounds and isocyanide: A facile entry to polysubstituted 2-cyanopyrroles
Yaping Zhang , Wei Zhou , Mingchun Gao , Tianqi Liu , Bingxin Liu , Chang-Hua Ding , Bin Xu

Polysubstituted pyrroles have attracted tremendous attention due to their wide existence in various bioactive natural products and pharmaceuticals. Among them, Tolmetin is a non-steroidal anti-inflammatory drug, which can be used in the treatment of rheumatoid arthritis [1]. Besides, Sunitinib is well acknowledged to promote tumor angiogenesis by inhibiting vascular endothelial growth factor receptor, further prolonging the median survival time of patients with advanced renal cell cancer [2]. Among polysubstituted pyrroles, 2-cyanopyrroles have proven to be privileged structure in medicinal chemistry, demonstrating many biological activities, including anti-inflammatory, anti-hyperlipidemia, hormones modulation, etc. (Fig. 1) [3,4]. For example, WAY-255348 exhibited potent and robust activity on progesterone receptor (PR) antagonist and contraceptive end points in the rat and rhesus monkey (Fig. 1) [5].
Owing to the significance of 2-cyanopyrroles in aspects of pharmacology and bioactivity, many efforts have been contributed to the skeleton construction. The most common method is the direct cyanation of pyrroles with a variety of cyanating reagents [6-17], including inorganic cyanides [7-9], chlorosulfonyl isocyanates [10,11], DMF [12,13], trimethylsilyl cyanide (TMSCN) [14,15] and tBuNC [16,17] (Scheme 1a). Rearrangement reactions from tetrazolo[1, 5-a]pyridines under pyrolysis could generate the 2-cyanopyrrole skeleton (Scheme 1b) [18,19]. Great progresses have been made through successive pyrrole synthesis and cyanation reaction, where two different "N" sources were required (Scheme 1c). For example, bimolecular tandem reactions were feasible by using α, β-unsaturated imines [20] or acrylnitriles [21] as the "N" source and TMSCN or tBuNC as the "CN" source for the 2-cyanopyrrole formation. Alternatively, multicomponent reactions were introduced by employing unsaturated olefins [22,23] or alkynes [24,25] as substrates. Among these examples, tBuNC was served as either "N" source or "CN" source. However, very few examples have been documented on the reactions involving isocyanides as the sole "N" and "CN" sources for the construction of 2-cyanopyrroles (Scheme 1d). One elegant example was illustrated by Inoue and co-workers from the reaction of acetylenes and tBuNC catalyzed by dinuclear palladium complexes, where isocyanide was also served as the palladium complex ligand [26]. Wang's group reported another method by the [3 + 2] cyclization of terminal alkynes with in-situ generated difluoroketenimine from difluorocarbene and tBuNC, providing fluorinated 2-cyanopyrroles [27]. A general and simple method is highly desirable for the synthesis of polysubstituted 2-cyanopyrroles through a new synthetic mode.

As critical building blocks, isocyanides have been widely applied in organic synthesis, medicinal chemistry, and materials science [28-36]. Isocyanide insertion into inert bonds under Lewis acid and transitional-metal catalysis proved to be an efficient approach for the construction of complicated molecules [37-42]. Nevertheless, the insertion of isocyanides into C(sp3)-H bonds was less studied due to the difficulty toward breaking inert C(sp3)-H bonds [39]. Therefore, the exploration of a facile method to 2-cyanopyrroles with the utilization of isocyanide insertion into C(sp3)-H bonds is greatly valuable. We have realized the isocyanide C(sp3)-H bond insertion reaction to afford pyrroles [43] and iminonitriles [44,45]. Very recently, a novel triple-consecutive isocyanide insertion with aldehydes was disclosed to afford 4-cyanooxazoles [46], where tBuNC serving as sources of critical "CN" and oxazole moieties. We envisioned this strategy could be applied to form other nitrile-containing heterocycles by the consecutive isocyanide insertion. Herein, we report a Brønsted acid-catalyzed sequential isocyanide insertion of allyl compounds, containing a heteroatom (N, O, and S) [47-49], to enable the expedient synthesis of 2-cyanopyrroles under mild conditions, where both pyrrole formation and cyanation reaction are realized via consecutive isocyanide insertion (Scheme 1d). The given 2-cyanopyrroles serve as versatile building blocks and could be transformed into other functionalized pyrroles containing primary amine, formyl, alkynyl, iodo or amide groups through postfunctionalizations. Furthermore, 2-cyanopyrrole could be used for the synthesis of nucleobase analogue of Remdesivir and 5H-pyrrolo[2, 1-a]isoindole, respectively.
Initially, we started our investigation by exploring the reaction of 1a with tert–butyl isocyanide at 80 ℃ in the presence of AgOTf and 2, 3-dichloro-5, 6-dicyano-1, 4-benzoquinone (DDQ) in chlorobenzene under a nitrogen atmosphere [44,45]. Intriguingly, an unexpected 2-cyanopyrrole product 3a was obtained in 68% yield, instead of the previous reported α-iminonitrile product (Table 1, entry 1) [44,45]. None or trace amounts of product 3a was afforded when DDQ was replaced by other oxidants including 1, 4-benzoquinone (BQ), oxone, tetrachloro-p-benzoquinone (p-chloranil) and [bis(trifluoroacetoxy)iodo]benzene (PIFA) (entries 2–5). When changing the usage of DDQ, the product 3a was obtained in decreased yield (entries 6 and 7). Further screening of solvent effect on the reaction indicated the use of DMF, DMSO, EtOAc, PhCN and dioxane resulted in no reaction or trace amount of product 3a (entries 8–12), while toluene and hexafluorobenzene (C6F6) gave lower yield than chlorobenzene (entries 13 and 14 vs. entry 1). Both atmosphere and temperature have certain influences on the reaction, since the yield decreased when the reaction was carried out under air or oxygen atmosphere or the temperature was varied (entries 15–18). Metal catalysts bearing a triflate anion were then tested, demonstrating favorable catalytic activities on the reaction (entries 19–21). Remarkably, the examination of Brønsted acid catalysts revealed that trifluoromethanesulfonic acid (TfOH) gave a slightly improved yield (72%) than AgOTf (entries 22–24 vs. entry 1). A control experiment indicated the reaction furnished 3a in 47% yield in the absence of TfOH, suggesting the importance of acid catalyst (entry 25).
With the optimized reaction conditions in hand, we then examined the substrate scope of 3-aryl allyl compounds and isocyanides. As summarized in Scheme 2, various substitution patterns and functional groups were well tolerated. 3-Aryl allylamines containing different substituents, such as alkoxy (3b), halides (3c, 3d, 3f) and alkyl (3e, 3g), were compatible with the reaction conditions, regardless of their different electronic properties and substituted positions. The structure of 3a was unequivocally determined by the single crystal X-ray diffraction analysis. Apart from monosubstituted phenyl allylamines, disubstituted phenyl allylamine could also afforded the desired product 3g in 69% yield. The reaction was not limited to allylamines containing substituted benzene ring. Substrates bearing different aromatic groups were transformed to the corresponding products smoothly as well, including naphthyl (3h), thienyl (3i), and even pyrenyl (3j) with large steric hindrance. The N-protecting groups of 3-aryl allylamines 1 were also investigated, which could be N-methylsulfonyl (Ms) (3k), N-nosyl (Ns) (3l), N-Ts-N-alkyl (3m–3p), N-Ts-N-Ph (3q), N-phthalimidyl (3r), and N-maleimidyl (3s), affording pyrrole products in moderate to high yields. Notably, when N, N-dicinnamyl-4-methyl-benzenesulfonamide was used as the substrate, only one 2-cyanopyrrole (3p) can be constructed, even though the amounts of isocyanide and DDQ have been doubled. Moreover, the adoption of (E)-4-methyl-N-(2-methyl-3-phenylallyl)benzenesulfonamide 1t as the substrate led to a fully substituted pyrrole (3t). Meanwhile, pyrrole product 3u could be obtained in 34% yield when 1, 1, 3, 3-tetramethylbutyl isocyanide was used instead of tert–butyl isocyanide. However, the attempt to use N-cinnamylacetamide or 4-cinnamylmorpholine as the reactant failed to afford the pyrroles under standard conditions, which might be attributed to the instability of in-situ formed iminium intermediates. In addition, when (E)-N-tosyl–but-2-en-1-amine was used as the substrate to react with 2a, no desired product was obtained. Also, when 1-adamantyl isocyanide or cyclohexyl isocyanide reacted with 1a, the reaction failed to afford desired product.

To further explore the generality of this protocol, a variety of allyl(thio)ethers were explored under the standard reaction conditions, as illustrated in Scheme 3. To our delight, the reaction also exhibited good functional group tolerance towards ether substrates 4. The variation of allyl ethers could affect the reaction with the yields decreased somewhat (5a–5g) compared with allylamines. It was noted that allyl ethers bearing an alkynyl or allyl group have two potential reaction sites, producing 5c and 5d selectively in 44% and 37% yields, respectively. A variety of allyl compounds with aryl or thienyl substituents could be converted to corresponding products (5h–5l). Similar to the 3-arylallylamine substrate, (E)-(3–methoxy-2-methylprop-1-en-1-yl)benzene 4m could afford fully substituted pyrrole (5m) in 45% yield. Noteworthily, for a diethylstilbestrol derivative, the desired product (5n) was produced in moderate yield. Subsequently, the use of pentadiene ether instead of allyl one could generate 5o in 40% yield. To further validate the feasibility of thioether in the reaction, thiopyrrole 5p could be achieved from benzyl(cinnamyl)sulfane 4p in moderate yield. It should be noted that the low yield of pyrrole products is due to the formation of some undeterminable by-products. Additionally, the reaction of cinnamaldehyde and tBuNC was carried out under the standard reaction conditions, but a complicated reaction was observed and no isolable product could be obtained.

In order to evaluate the practicality of the given method, a gram-scale synthesis was performed to afford the product 3a in 73% yield under the standard conditions (Scheme 4a). The access to molecular diversity was demonstrated by the post-functionalizations of generated pyrrole products 3a and 5a, as shown in Scheme 4. Multiple transformations of substituents on 3a could proceed smoothly and various functional group including primary amine, formyl, alkynyl, iodo and amide were introduced successfully (Scheme 4a). These functionalized pyrroles were not easily accessible through usual approach and will find potential applications for further deliberation, generating advanced organic building blocks. Among which, the isolation of product 6 could be achieved by the cleavage of N-sulfonamide group of 3a in the presence of Ti(OiPr)4, TMSCl and Mg powder. Alternatively, the primary amine 6 could be afforded in quantitative yield through the deprotection of N-phthalimidyl group of pyrrole 3r in the presence of hydrazine hydrate. The N–tert–butyl substituent was removed smoothly with the assistance of hydrochloric acid, affording product 7 in almost quantitative yield. Besides, the selective conversion of cyano group of 3a could be realized under different conditions, providing 2-formyl pyrrole 8 and 2-carboxamidyl pyrrole 9 in good yields, respectively. The iodo group was introduced into 3a smoothly by reacting with N-iodosuccinimide and gave a fully substituted pyrrole 10, which was subsequently reacted with trimethylsilylacetylene through the Sonogashira coupling. Followed by the removal of trimethylsilyl (TMS) group, the product 12 with a terminal alkynyl group was afforded successfully. In addition, through the introduction of bromo group and Suzuki coupling with phenylboronic acid, 5a was transformed into a 4, 5-diphenylpyrrole 14 in 50% yield (Scheme 4b), which was the key precursor of bifunctional antitumor agents [50]. Notably, after the removal of N–tert–butyl group of 5a in the presence of TfOH, compound 15 was successfully ammoniated with the utilization of O-(2, 4-dinitrophenyl)hydroxylamine and NaH [51], and then cyclized with formamidine acetate to produce pyrrolo[2, 1-f][1, 2, 4]triazine 17 in 79% yield [52], which is the analogue of the nucleobase of Remdesivir, one of the most promising anti-COVID-19 medicines at present. Besides, with the promotion of [Cp*RhCl2]2 and Cu(OAc)2, the cyclization of compound 15 could occur successfully with ethyl acrylate, leading to the formation of 5H-pyrrolo[2, 1-a]isoindole 18 in 83% isolated yield (Scheme 4b) [53]. All of the above transformations demonstrated that this protocol possessed extraordinary reactivity towards various regents, allowing the efficient assembly of diverse pyrrole derivatives and further application in multiple fronts.

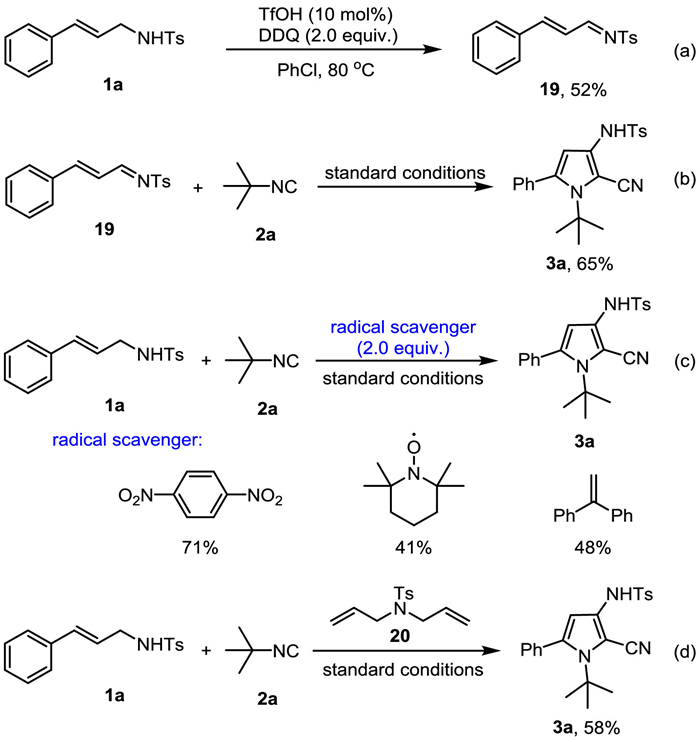
To gain insight into the possible intermediates and pathway of this reaction, control experiments were performed (Scheme 5). When 1a was subjected to the reaction in the absence of tert–butyl isocyanide, an imine compound 19 could be isolated in 52% yield (Scheme 5a), which could deliver the final product 3a in 65% yield under standard reaction conditions, indicating that the imine 19 might be an intermediate of this reaction (Scheme 5b). Various radical scavengers were then tried (Scheme 5c). The reaction occurred smoothly with 1, 4-dinitrobenzene as radical inhibitor and no significant decrease of yield was observed, while addition of 2, 2, 6, 6-tetramethylpiperidoxyl (TEMPO) and ethene-1, 1-diyldibenzene led to a moderate yield of product 3a. No cyclization product was observed from the radical clock probe 20, along with the formation of 3a in 58% yield (Scheme 5d). These above experimental results suggested that the radical process might be ruled out for this reaction at the current stage.
Although a detailed reaction pathway remains to be clarified, a plausible mechanism for this reaction was proposed on the basis of the above results, as depicted in Scheme 6. Initially, allyl compound 1a was oxidized by DDQ to form an iminium intermediate A [54,55] or α, β-unsaturated imine compound 19, while the iminium ion A could be further generated from the imine 19 in the presence of TfOH. Further nucleophilic attack by isocyanide to the iminium A afforded an intermediate B. Subsequently, the second nucleophilic attack by another isocyanide molecule on the intermediate B led to an intermediate C, which could be converted into the intermediate D through the expulsion of isobutene, along with the regeneration of TfOH to complete the catalytic cycle. Intermediate D was oxidized by DDQ again to generate another iminium intermediate E. Ultimately, the desired product 3a might be produced through the Nazarov-type cyclization [56] of E and the following aromatization of F.

In summary, a facile oxidative isocyanide insertion was disclosed for the modular synthesis of 2-cyanopyrroles from easily available allyl compounds. The given protocol featured a new formal [3 + 2] mode for pyrrole synthesis involving the regioselective isocyanide insertion to the C(sp3)–H bond and utilizing isocyanide as sources of critical "CN" and pyrrole moieties. Generally, broad substrate scope, good functional group tolerance, and operational simplicity enabled this approach promising and attractive for organic synthesis. The 2-cyanopyrrole products have been demonstrated as versatile synthons and were converted into nucleobase analogue of Remdesivir and 5H-pyrrolo[2, 1-a]isoindole. Further application of this method is underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We thank the National Natural Science Foundation of China (Nos. 22171178, 21871174, 22071143) and Innovation Program of Shanghai Municipal Education Commission (No. 2019–01–07–00–09-E00008) for financial support. The authors thank Prof. Qitao Tan (SHU) for helpful discussion.
M.L. Hyneck, P.C. Smith, A. Munafo, A.F. McDonagh, L.Z. Benet, Clin. Pharmacol. Ther. 44 (1988) 107–114. doi: 10.1038/clpt.1988.120
M.N. Paludetto, C. Bijani, F. Puisset, et al., J. Med. Chem. 61 (2018) 7849–7860. doi: 10.1021/acs.jmedchem.8b00812
L.D. Bratton, B. Auerbach, C. Choi, et al., Bioorg. Med. Chem. 15 (2007) 5576–5589. doi: 10.1016/j.bmc.2007.05.031
J. Bullington, D. Argentieri, K. Averill, et al., Bioorg. Med. Chem. Lett. 16 (2006) 6102–6106. doi: 10.1016/j.bmcl.2006.08.101
A. Fensome, W.R. Adams, A.L. Adams, et al., J. Med. Chem. 51 (2008) 1861–1873. doi: 10.1021/jm701080t
S. Pimparkar, A. Koodan, S. Maiti, et al., Chem. Commun. 57 (2021) 2210–2232. doi: 10.1039/d0cc07783f
W. Liu, Y. Ma, Y.F. Zhao, Y.W. Yin, J. Heterocycl. Chem. 43 (2006) 681–684. doi: 10.1002/jhet.5570430322
G. Cirrincione, A.M. Almerico, A. Passannanti, P. Diana, F. Mingoia, Synthesis (1997) 1169–1173 1997. doi: 10.1055/s-1997-1326
X. Teng, H. Keys, J. Yuan, A. Degterev, G.D. Cuny, Bioorg. Med. Chem. Lett. 18 (2008) 3219–3223. doi: 10.1016/j.bmcl.2008.04.048
G.H. Barnett, H.J. Anderson, C.E. Loader, Can. J. Chem. 58 (1980) 409–411. doi: 10.1139/v80-065
C.E. Loader, H.J. Anderson, Can. J. Chem. 59 (1981) 2673–2683. doi: 10.1139/v81-384
D.J. Paymode, F.S.P. Cardoso, T. Agrawal, et al., Org. Lett. 22 (2020) 7656–7661. doi: 10.1021/acs.orglett.0c02848
S. Ushijima, H. Togo, Synlett (2010) 1067–1070.
T. Dohi, K. Morimoto, Y. Kiyono, H. Tohma, Y. Kita, Org. Lett. 7 (2005) 537–540. doi: 10.1021/ol0476826
T. Dohi, K. Morimoto, N. Takenaga, et al., J. Org. Chem. 72 (2007) 109–116. doi: 10.1021/jo061820i
S. Xu, X. Huang, X. Hong, B. Xu, Org. Lett. 14 (2012) 4614–4617. doi: 10.1021/ol302070t
X. Hong, H. Wang, G. Qian, Q. Tan, B. Xu, J. Org. Chem. 79 (2014) 3228–3237. doi: 10.1021/jo500087g
C. Wentrup, H.W. Winter, J. Am. Chem. Soc. 102 (1980) 6159–6161. doi: 10.1021/ja00539a039
R.A. Evans, C. Wentrup, J. Chem. Soc., Chem. Commun. (1992) 1062–1064.
E. Marchand, G. Morel, S. Sinbandhit, Eur. J. Org. Chem. 1999 (1999) 1729–1738.
S.K. Guchhait, S. Sisodiya, M. Saini, et al., J. Org. Chem. 83 (2018) 5807–5815. doi: 10.1021/acs.joc.8b00465
Q.W. Gui, F. Teng, Z.C. Li, et al., Org. Chem. Front. 7 (2020) 4026–4030. doi: 10.1039/d0qo01113d
Q.W. Gui, F. Teng, S.N. Ying, et al., Chin. Chem. Lett. 31 (2020) 3241–3244. doi: 10.1016/j.cclet.2020.07.017
X.Q. Mou, Z.L. Xu, L. Xu, et al., Org. Lett. 18 (2016) 4032–4035. doi: 10.1021/acs.orglett.6b01883
Q.W. Gui, X. He, W. Wang, et al., Green Chem. 22 (2020) 118–122. doi: 10.1039/c9gc02657f
N. Tsukada, M. Wada, N. Takahashi, Y. Inoue, J. Organomet. Chem. 694 (2009) 1333–1338. doi: 10.1016/j.jorganchem.2008.12.039
R. Zhang, Z. Zhang, K. Wang, J. Wang, J. Org. Chem. 85 (2020) 9791–9800. doi: 10.1021/acs.joc.0c01120
G. Qiu, Q. Ding, J. Wu, Chem. Soc. Rev. 42 (2013) 5257–5269. doi: 10.1039/c3cs35507a
V.P. Boyarskiy, N.A. Bokach, K.V. Luzyanin, V.Y. Kukushkin, Chem. Rev. 115 (2015) 2698–2779. doi: 10.1021/cr500380d
T.R. Roose, D.S. Verdoorn, P. Mampuys, et al., Chem. Soc. Rev. 51 (2022) 5842–5877. doi: 10.1039/d1cs00305d
D. Chen, Y. Shan, J. Li, et al., Org. Lett. 21 (2019) 4044–4048. doi: 10.1021/acs.orglett.9b01220
Z. Liu, S. Cao, J. Wu, et al., ACS Catal. 10 (2020) 12881–12887. doi: 10.1021/acscatal.0c02867
S. Cheng, Y. Luo, T. Yu, et al., ACS Catal. 12 (2022) 837–845. doi: 10.1021/acscatal.1c05319
T. Yu, Z.Q. Li, J. Li, et al., ACS Catal. 12 (2022) 13034–13041. doi: 10.1021/acscatal.2c04461
W.B. Cao, J.D. Zhang, M.M. Xu, et al., Org. Lett. 24 (2022) 4620–4624. doi: 10.1021/acs.orglett.2c01736
S. Ren, K. Huang, J.B. Liu, et al., Chin. Chem. Lett. 33 (2022) 4870–4873. doi: 10.1016/j.cclet.2022.02.028
H. Wang, B. Xu, Chin. J. Org. Chem. 35 (2015) 588–602. doi: 10.6023/cjoc201411035
B. Song, B. Xu, Chem. Soc. Rev. 46 (2017) 1103–1123. doi: 10.1039/C6CS00384B
W. Wang, T. Liu, C.H. Ding, B. Xu, Org. Chem. Front. 8 (2021) 3525–3542. doi: 10.1039/d1qo00153a
Z. Guan, S. Zhu, Y. Yang, et al., Chem. Sci. 12 (2021) 14121–14125. doi: 10.1039/d1sc04475c
Y.M. Zhu, Y. Fang, H. Li, X.P. Xu, S.J. Ji, Org. Lett. 23 (2021) 7342–7347. doi: 10.1021/acs.orglett.1c02422
Y. Shan, L. Su, D. Chen, et al., Chin. Chem. Lett. 32 (2021) 437–440. doi: 10.1016/j.cclet.2020.04.041
Z. Tian, J. Xu, B. Liu, Q. Tan, B. Xu, Org. Lett. 20 (2018) 2603–2606. doi: 10.1021/acs.orglett.8b00798
H. Chi, H. Li, B. Liu, et al., iScience 21 (2019) 650–663. doi: 10.1016/j.isci.2019.10.057
L. Zhao, B. Liu, Q. Tan, C.H. Ding, B. Xu, Org. Lett. 21 (2019) 9223–9227. doi: 10.1021/acs.orglett.9b03590
S. Lu, C.H. Ding, B. Xu, Org. Lett. 25 (2023) 849–854. doi: 10.1021/acs.orglett.3c00008
W. Zhao, J. Sun, Chem. Rev. 118 (2018) 10349–10392. doi: 10.1021/acs.chemrev.8b00279
Z.L. Xia, Q.F. Xu-Xu, C. Zheng, S.L. You, Chem. Soc. Rev. 49 (2020) 286–300. doi: 10.1039/c8cs00436f
Y.B. Chen, P.C. Qian, L.W. Ye, Chem. Soc. Rev. 49 (2020) 8897–8909. doi: 10.1039/d0cs00474j
G.B. Jones, J.E. Mathews, Bioorg. Med. Chem. Lett. 7 (1997) 745–748. doi: 10.1016/S0960-894X(97)00098-X
Z. Chen, S.H. Kim, S.A. Barbosa, et al., Bioorg. Med. Chem. Lett. 16 (2006) 628–632. doi: 10.1016/j.bmcl.2005.10.052
S.A. Patil, B.A. Otter, R.S. Klein, J. Heterocycl. Chem. 31 (1994) 781–786. doi: 10.1002/jhet.5570310415
Q. Han, X. Guo, Z. Tang, et al., Adv. Synth. Catal. 360 (2018) 972–984. doi: 10.1002/adsc.201701381
J.L. Miller, J.M.I.A. Lawrence, F.O. Rodriguez del Rey, P.E. Floreancig, Chem. Soc. Rev. 51 (2022) 5660–5690. doi: 10.1039/d1cs01169c
J.L. Miller, L. Zhou, P. Liu, P.E. Floreancig, Chem. Eur. J. 28 (2022) e202103078. doi: 10.1002/chem.202103078
H. Pellissier, Tetrahedron 61 (2005) 6479–6517. doi: 10.1016/j.tet.2005.04.014

Scheme 2 Substrate scope of allylamines. Reaction conditions: 1 (0.3 mmol), DDQ (0.6 mmol), tBuNC (0.9 mmol), TfOH (0.03 mmol), and PhCl (1.5 mL), 80 ℃, 3 h, under nitrogen atmosphere. Yields shown are of the isolated products. a DDQ (0.9 mmol), tBuNC (0.9 mmol). b DDQ (1.2 mmol), tBuNC (1.8 mmol), TfOH (0.06 mmol) and PhCl (1.5 mL).

Scheme 3 Substrate scope of allyl(thio)ethers. Reaction conditions: 4 (0.3 mmol), DDQ (0.6 mmol), tBuNC (0.9 mmol), TfOH (0.03 mmol) and PhCl (1.5 mL), 80 ℃, 3 h, under nitrogen atmosphere. Yields shown are of the isolated products. a DDQ (0.6 mmol), tBuNC (0.9 mmol), AgOTf (0.03 mmol) and PhCl (1.5 mL). b 4n (0.2 mmol), DDQ (0.4 mmol), tBuNC (0.6 mmol), TfOH (0.02 mmol) and PhCl (1.5 mL).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:




 下载:
下载: