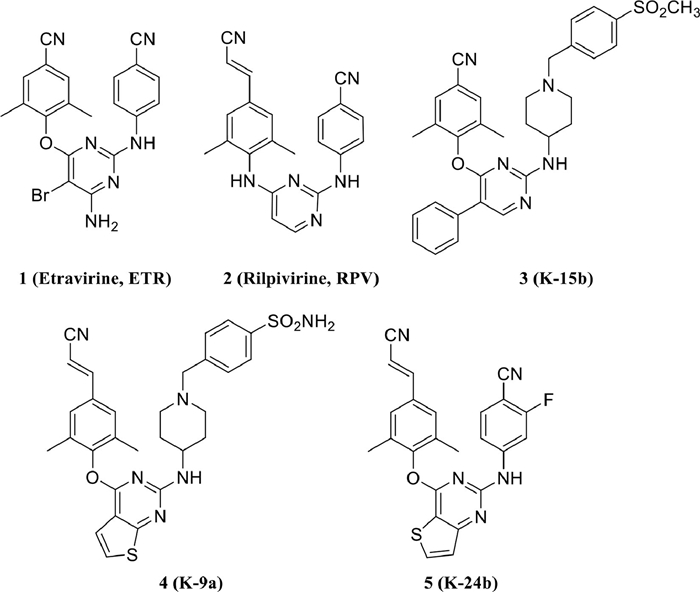
Figure 1.
Chemical structures of ETR, RPV and our previously reported NNRTIs 3, 4, and 5.
Design, synthesis, and biological evaluation of benzo[4,5]thieno[2,3-d]pyrimidine derivatives as novel HIV-1 NNRTIs
Bairu Meng , Zongji Zhuo , Han Yu , Sining Tao , Zixuan Chen , Erik De Clercq , Christophe Pannecouque , Dongwei Kang , Peng Zhan , Xinyong Liu

The acquired immune deficiency syndrome (AIDS), a malignant infectious disease caused by human immunodeficiency virus (HIV), has affected nearly 38.4 million patients with 0.65 million deaths at the end of 2021 [1]. HIV is a member of the Retroviridae family and includes two subtypes: HIV-1 and HIV-2. Between them, HIV-1 is the main causative agent of AIDS on account of its higher infectivity and pathogenicity [2]. During the HIV-1 replication cycle, reverse transcriptase (RT) is responsible for the conversion of the viral single-stranded RNA into double-stranded proviral DNA [3]. Without RT, replication is impaired and the viral life cycle is shut down [4, 5]. Thus, RT has garnered significant attention and RT inhibitors are key components of the highly active antiretroviral therapy (HARRT) regimen [6]. Currently, there are two types of RT inhibitors, including nucleoside RT inhibitors (NRTIs) which are incorporated into the elongating viral DNA and prevent further elongation of the growing DNA strand and non-nucleoside RT inhibitors (NNRTIs) which bind to an allosteric pocket (NNRTI-binding pocket, NNIBP) near but distinct from the active site to inhibit RT functional status [7]. NNRTIs have shown promising selectivity and drug-like properties in contrast to NRTIs [8-12]. Up to now, six HIV-1 NNRTIs drugs have been approved by U.S. Food and Drug Administration (FDA), including the first-generation drugs delavirdine (DLV), nevirapine (NVP), and efavirenz (EFV) and the second-generation drugs etravirine (ETR), rilpivirine (RPV), and doravirine (DOR) [13]. Moreover, dapivirine (DPV, TMC120) was approved by the European Medicines Agency for marketing in 2020 [14]. However, the single mutants K103N and Y181C seriously reduced the effectiveness of the first-generation NNRTIs [15, 16]. Although the second-generation NNRTIs displayed improved activity against these single mutants, adverse effects and the newly emerging single mutation (E138K) and double mutation RES056 (K103N + Y181C) severely limited their clinical application [17]. Therefore, there is an urgent need to discover new generation of drugs with more effective activity and safety profiles.
With ETR as the lead compound, series of novel potent HIV-1 NNRTIs have been identified in our lab, such as 2,4,5-trisubstituted pyrimidine derivative 3 [18], piperidine-substituted thiophene[2,3-d]pyrimidine derivative 4 [19] and fluorine-substituted diaryl-thiophene[3,2-d]pyrimidine derivative 5 (Fig. 1) [20]. Among them, 4 and 5 were demonstrated with highly potent antiviral activity and improved resistance profiles against HIV-1 wild-type (WT) and a panel of NNRTIs-resistant strains. Although 3 showed an outstanding activity against HIV-1 WT with a 50% effective concentration (EC50) value of 4.93 nmol/L, its potency toward the most common double-mutant strain RES056 (EC50 = 590 nmol/L) sharply decreased. The conformational superposition analysis of 3 and 4 with RT indicated that they adopted a similar horseshoe-like conformation in the NNIBP. The binding modes of 4 illustrated that the S atom of thiophene[2,3-d]pyrimidine scaffold was involved in a hydrogen-bond with the backbone of Lys101 through a bridging water molecule, which were believed to play a crucial role in improving resistance to drug resistance. In the case of compound 3, its phenyl group which linked to the C5 position of the central pyrimidines can point to the tolerant region II of NNIBP, but it did not develop any interactions with NNIBP. Therefore, given the results obtained from our previous explorations, it was still worth further exploring the elaborated structural modifications of central core.
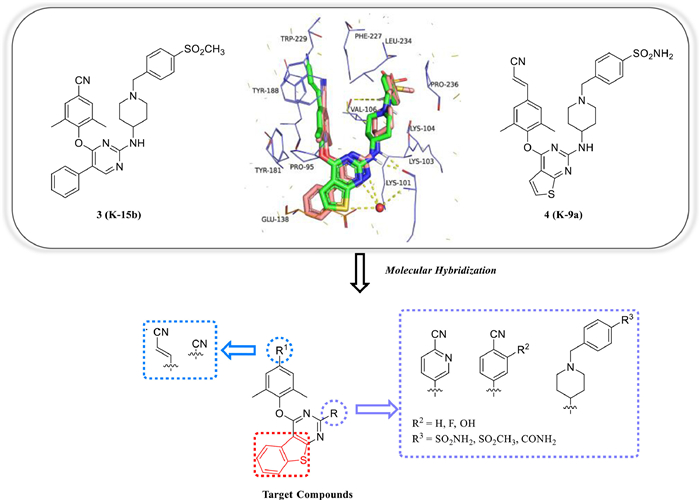
The binding modes of 3 and 4 with WT HIV-1 RT revealed that the central pyrimidine fit in the same site and the thienyl substitution of 4 blocked in between the 5-phenylpyrimidine of 3, suggesting that we could merge the two central cores into the benzo[4,5]thieno[2,3-d]pyrimidine (Fig. 2). We hope the newly introduced central core could establish more effective interactions with Lys101 to exhibit improved drug resistance profiles and get a deeper insight of the structure−activity relationships (SARs) of the NNIBP tolerant region II. Additionally, the privileged left wings of 3 and 4 were kept in this work. Meanwhile, the cyanophenyl groups and piperidine-linked aryl structures in the right wing of 4 and 5 were introduced to the benzo[4,5]thieno[2,3-d]pyrimidine scaffold via molecular hybridization and bioisosterism strategies. A totally of 14 novel derivatives were designed, synthesized and performed their biological evaluation in this work.
As depicted in Scheme 1, ethyl 2-aminobenzo[b]thiophene-3-carboxylate and urea were used as the starting materials to synthesis intermediate 7 via microwave reaction at 200 ℃, and intermediate 7 was subsequently treated with POCl3 to yield the central benzo[4,5]thieno[2,3-d]pyrimidine scaffold 8. Compound 9 was obtained by substitution reaction of 8 with 4-hydroxy-3,5-dimethylbenzonitrile in the presence of potassium carbonate (K2CO3) in dimethylformamide (DMF) at room temperature (r.t.). Likewise, 8 was reacted with (E)−3-(4-hydroxy-3,5-dimethylphenyl)acrylonitrile to afford intermediate 10.

As depicted in Scheme 2, Pd-catalyzed Buchwald-Hartwig coupling of 9 or 10 with various cyano aromatic amines yielded 11a–d and 12a–d. Compound 9 or 10 was treated with N-(tert-butoxycarbonyl)−4-amino-piperidine and subsequent deprotection delivered 13 or 14 using trifluoroacetic acid (TFA), which was reacted with substituted benzyl chloride (or bromide) to give the target compounds 15a–c and 16a–c. Detailed procedures and compound characterizations can be found in Supporting information.

All the newly synthesized compounds were evaluated for their biological activity to WT HIV-1 IIIB strain and a double-mutant HIV-1 strain RES056 (K103N/Y181C) in MT-4 cells with the MTT method. The selected potent inhibitors were tested for their activity against other common NNRTIs-resistant strains, including L100I, K103N, Y181C, Y188L, E138K, and F227L/V106A. NVP, ETR, and EFV were acted as reference drugs. Experimental procedure can be found in Supporting information. The values of EC50 (anti-HIV-1 potency), 50% cytotoxicity concentration (CC50), selectivity index (SI, CC50/EC50 ratio) of these novel inhibitors were determined and shown in Tables 1 and 2.
 DownLoad:
CSV
DownLoad:
CSV

|
DownLoad:
CSV

|
As depicted in Table 1, most compounds exhibited promising potency against HIV-1 IIIB with EC50 values of 0.021 µmol/L to 0.100 µmol/L. Of special note is that 16b with the -SO2CH3 group showed the highest activity against HIV-1 IIIB (EC50 = 0.021 µmol/L) and outstanding activity against RES056 (EC50 = 0.298 µmol/L). Although 16b displayed slightly inferior antiviral activities compared to ETR (EC50 IIIB = 0.003 µmol/L, EC50 RES056 = 0.083 µmol/L), it demonstrated much reduced cytotoxicity (CC50 > 200 µmol/L) and higher SI values (SIIIIB > 9419, SIRES056 > 672) than that of ETR (CC50 > 4.59 µmol/L, SIIIIB > 1633, SIRES056 > 55). Replacing the -SO2CH3 group in the R2 position of 16b with -SO2NH2 group and -CO2NH2 group resulted in compounds 16a and 16c, which showed promising activity against HIV-1 IIIB (EC50 = 0.027 and 0.100 µmol/L, respectively). Interestingly, changing piperidine-linked substituted benzene groups to the cyano aromatic amines of 12a, 12b, and 12d (EC50 IIIB = 0.023–0.085 µmol/L, EC50 RES056 = 0.565–0.960 µmol/L) also showed considerable activity against HIV-1 IIIB but all of their potencies against RES056 still did not exceed 16b and ETR. Particularly, 12d (EC50 IIIB = 0.023 µmol/L, EC50 RES056 = 0.894 µmol/L) exhibited comparable activity against HIV-1 IIIB but inferior activity against RES056, being comparable to that of ETR. However, 12c was devoid of activity against HIV-1 IIIB and RES056, suggesting that cyano aromatic structure with a 2-hydroxy group in the right wing had an adverse effect on the potency of compounds against a panel of NNRTI-resistant strains. When replacing the cyanovinyl substituent with cyano substituent on the left wing, several compounds 11a (EC50 = 0.041 µmol/L), 15a (EC50 = 0.030 µmol/L) and 15b (EC50 = 0.035 µmol/L) retained potency against HIV-1 IIIB. But they showed inferior potency against RES056, declaring that cyanovinyl substituent was important to improve resistance profiles.
Additionally, 11a, 12a, 12d, 15a, 15b, 16a and 16b were selected to performed their antiviral potency against a panel of single mutant HIV-1 strains, including L100I, K103N, Y181C, Y188L, E138K, and double mutant strain F227L + V106A. As shown in Table 2, the most potent RES056 inhibitor 16b could strongly inhibit these mutant strains, with EC50 values of 0.027 µmol/L (L100I), 0.017 µmol/L (K103N), 0.085 µmol/L (Y181C), 0.121 µmol/L (Y188L), 0.112 µmol/L (E138K), and 0.041 µmol/L (F227L + V106A). In addition, 16a also showed potent inhibitory activity against L100I (EC50 = 0.029 µmol/L), K103N (EC50 = 0.026 µmol/L), and F227L + V106A (EC50 = 0.043 µmol/L), while showing weak activity against Y181C (EC50 = 0.119 µmol/L), Y188L (EC50 = 0.121 µmol/L), and E138K (EC50 = 0.175 µmol/L). These results demonstrated that the structure of the cyano-group on the left and the aminopyridine on the right was more conducive to the improvement of the activity of the compound against the mutant strains, and compound 16b with these two structural features exhibited the optimal resistance to drug resistance.
The ability of representative compounds to inhibit recombinant WT HIV-1 RT enzyme were measured in vitro to confirm the binding target of these novel inhibitors. NVP and ETR were chosen as the reference drugs in this assay. Detailed procedures can be found in Supporting information. As depicted in Table 3, these compounds exhibited significant binding-affinity to WT HIV-1 RT (IC50 = 0.099–0.366 µmol/L), being about 3−10-fold potent than that of NVP (IC50 = 1.02 µmol/L). Although these novel inhibitors exhibited inferior activities compared to that of ETR (IC50 = 0.011 µmol/L), these results could validate that the binding target of the newly designed compounds was HIV-1 RT.
DownLoad:
CSV

|
To elucidate the contribution of the 2,4-bisubstituted benzo[4,5]thieno[2,3-d]pyrimidine motif in 16b binding to WT HIV-1 RT (from PDB ID: pdb:6C0N) and RES056 HIV-1 RT (from PDB ID: pdb:6C0R), molecular docking study was performed with Maestro (Maestro, Schrödinger, LLC, New York, NY, 2019). The docking results were visualized by PyMOL. Detailed procedures can be found in Supporting information. As shown in Fig. 3, 16b adopted a horseshoe conformation in the NNIBP consistent with classical DAPY derivatives. Concretely, the docking results maintained the identify of our previous investigations and could be described as follows: (1) Benzothiophene group of the central core sat in the tolerant region II of NNIBP as envisioned and the thiophene S atom formed a hydrogen bond with Lys101 through a bridging water molecule, which provided great benefits to inhibit RT and the N atom of the pyrimidine also formed a hydrogen bond with the backbone of Lys101; (2) The left wing fully occupied the hydrophobic sub-pocket and developed stronger hydrophobic interactions with Phe227 and Trp229; (3) The right wing also showed similar interactions with Val106 as in other RT/DAPY structures. It was worth mentioning that 16b lost essential water-mediated hydrogen bonds with Lys101, making it reduced potency that of 3 and 4.
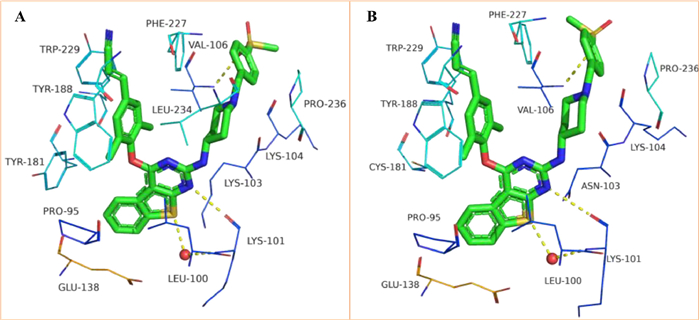
We next predicted the binding mode of 16b with RES056 RT. Due to the flexibility of the right wing of 16b, the K103N mutation did not affect resistance profiles. However, the mutation of Tyr181 residue (Y181C) locating in the hydrophobic tunnel had an adverse effect on the hydrophobic interactions between left wing and Tyr181. The molecular planarity of the fused benzo[4,5]thieno[2,3-d]pyrimidine scaffold may also account for the decreased resistance profiles of 16b.
In the current study, we leverage our understanding of the HIV-1 RT inhibition mechanism based on the binding modes of HIV-1 RT with two inhibitors to guide the lead optimization. Based on the superposition analysis of co-crystal structures, the central cores of 3 and 4 were merged into the benzo[4,5]thieno[2,3-d]pyrimidine to improve compounds' antiviral activity against WT and mutant HIV-1 strains. The results demonstrated that 16b exhibited the most potent activity against HIV-1 WT strain and NNRTIs-resistant strains (EC50 = 0.017–0.298 µmol/L). Moreover, it also showed significant low cytotoxicity (CC50 > 200 µmol/L) and high SI values. The enzymatic inhibitory activity (IC50 = 0.183 µmol/L) results proved the target of these novel compounds were HIV-1 RT. The docking results verified that the N atom and S atom of the central core could form double hydrogen bonds with the backbone of Lys101, which were in agreement with our expectation. Taken together, 16b was a novel HIV-1 RT inhibitor that displayed potent antiviral activity and thus had promising potential for further study.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (NSFC, Nos. 81973181, 82273773), Shandong Provincial Natural Science Foundation (Nos. ZR2020YQ61, ZR2020JQ31), Qilu Young Scholars Program of Shandong University and Taishan Scholar Program at Shandong Province. The technical assistance of Mr. Kris Uyttersprot and Mrs. Kristien Erven, for the HIV experiments is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
HIV data and statistics.
A.S. Fauci, H.C. Lane, N. Engl. J. Med. 383 (2020) 1–4. doi: 10.1056/NEJMp1916753
G. Bec, B. Meyer, M.A. Gerard, et al., J. Am. Chem. Soc. 135 (2013) 9743–9752. doi: 10.1021/ja4018418
A. Tavassoli, Chem. Soc. Rev. 40 (2011) 1337–1346. doi: 10.1039/C0CS00092B
C. Flexner, Nat. Rev. Drug Discov. 6 (2007) 959–966. doi: 10.1038/nrd2336
R.J. Shattock, M. Warren, S. McCormack, C.A. Hankins, Science 333 (2011) 42–43. doi: 10.1126/science.1206399
D. Li, P. Zhan, E. De Clercq, X. Liu, J. Med. Chem. 55 (2012) 3595–3613. doi: 10.1021/jm200990c
D. Feng, F. Wei, Y. Sun, et al., Chin. Chem. Lett. 32 (2021) 4053–4057. doi: 10.1016/j.cclet.2021.02.033
D. Feng, H. Lin, L. Jiang, et al., Eur. J. Med. Chem. 246 (2023) 114957. doi: 10.1016/j.ejmech.2022.114957
F. Zhao, H. Zhang, M. Xie, et al., J. Med. Chem. 66 (2023) 2102–2115. doi: 10.1021/acs.jmedchem.2c01875
Z. Wang, D. Kang, D. Feng, et al., Eur. J. Med. Chem. 206 (2020) 112811. doi: 10.1016/j.ejmech.2020.112811
S. Han, Y. Lei, C. Pannecouque, et al., Chin. Chem. Lett. 31 (2020) 764–768. doi: 10.1016/j.cclet.2019.11.020
Z. Wang, S. Cherukupalli, M. Xie, et al., J. Med. Chem. 65 (2022) 3729–3757. doi: 10.1021/acs.jmedchem.1c01758
N. Bhavaraju, K. Shears, K. Schwartz, et al., Curr. HIV AIDS Rep. 18 (2021) 508–517. doi: 10.1007/s11904-021-00577-8
D.A. Lehman, D.C. Wamalwa, C.O. McCoy, et al., J. Acquir. Immune. Defic. Syndr. 60 (2012) 225–233. doi: 10.1097/QAI.0b013e3182515730
M.E. Cilento, K.A. Kirby, S.G. Sarafianos, Chem. Rev. 121 (2021) 3271–3296. doi: 10.1021/acs.chemrev.0c00967
A. Olson, N. Bannert, A. Sönnerborg, et al., AIDS 32 (2018) 161–169. doi: 10.1097/QAD.0000000000001689
D. Kang, F.X. Ruiz, Y. Sun, et al., J. Med. Chem. 64 (2021) 4239–4256. doi: 10.1021/acs.jmedchem.1c00268
D. Kang, D. Feng, Y. Sun, et al., J. Med. Chem. 63 (2020) 4837–4848. doi: 10.1021/acs.jmedchem.0c00117
D. Kang, F.X. Ruiz, D. Feng, et al., J. Med. Chem. 63 (2020) 1298–1312. doi: 10.1021/acs.jmedchem.9b01769
D. Kang, H. Zhang, Z. Wang, et al., J. Med. Chem. 62 (2019) 1484–1501. doi: 10.1021/acs.jmedchem.8b01656

Figure 1 Chemical structures of ETR, RPV and our previously reported NNRTIs 3, 4, and 5.

Scheme 1 Synthesis of 9 and 10. Reagents and conditions: (ⅰ) urea, microwave reaction, 200 ℃; (ⅱ) POCl3, 110 ℃; (ⅲ) K2CO3, DMF, r.t.

Scheme 2 Synthesis of 11a–d, 12a–d, 15a–c and 16a–c. Reagents and conditions: (ⅰ) BINAP, PdCl2(PPh3)2, Cs2CO3, 1,4-dioxane, 120 ℃; (ⅱ) K2CO3, DMF, reflux; then TFA, DCM, r.t.; (ⅲ) substituted benzyl chloride (or bromide), K2CO3, DMF, r.t.

Figure 3 Predicted binding modes of 16b with the HIV-1 WT RT (A, PDB code: 6C0N) and RES056 HIV-1 RT (B, PDB code: 6C0R).
Table 1. Antiviral potency against HIV-1 IIIB and RES056, cytotoxicity, and SI values of target compounds 11a–d, 12a–d, 15a–c and 16a–c.
|
|
 下载: 导出CSV
下载: 导出CSV
Table 2. Antiviral potency against mutant HIV-1 strains of target compounds 11a, 12a, 12d, 15a, 15b, 16a and 16b.
|
|
下载: 导出CSV
Table 3. Inhibitory activity against WT HIV-1 RT of target compounds 11a, 12a, 12d, 15a, 15b, 16a and 16b.
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
