Received Date:
01 June 2023 Accepted Date:
19 July 2023 Revised Date:
17 July 2023 Available Online:
15 May 2024
Abstract:
Strand displacement reaction is a crucial component in the assembly of diverse DNA-based nanodevices, with the toehold-mediated strand displacement reaction representing the prevailing strategy. However, the single-stranded Watson-Crick sticky region that serves as the trigger for strand displacement can also cause leakage reactions by introducing crosstalk in complex DNA circuits. Here, we proposed the toeless and reversible DNA strand displacement reaction based on the Hoogsteen-bond triplex, which is compatible with most of the existing DNA circuits. We demonstrated that our proposed reaction can occur at pH 5 and can be reversed at pH 9. We also observed an approximately linear relationship between the degree of reaction and pH within the range of pH 5–6, providing the potential for precise regulation of the reaction. Meanwhile, by altering the sequence orientation, we have demonstrated that our proposed reaction can be initiated or regulated through the same toeless mechanism without the requirement for protonation in low pH conditions. Based on the proposed reaction principle, we further constructed a variety of DNA nanodevices, including two types of DNA logic gates that rely on pH 5/pH 9 changes for initiating and reversing: the AND gate and the OR gate. We also successfully constructed a DNA Walker based on our proposed reaction modes, which can move along a given track after the introduction of a programmable DNA sequence and complete a cycle after 4 steps. Our findings suggest that this innovative approach will have broad utility in the development of DNA circuits, molecular sensors, and other complex biological systems.
DNA is a highly programmable biomolecule, which can be designed into complex DNA nanostructures based on the specificity of its base pairing through Watson-Crick bonds, such as DNA logic circuits [1-3], various nanomachines [4-6], and biosensors [7-10]. Toehold-mediated DNA strand displacement reactions are the driving force for the majority of reactions in the field of dynamic DNA nanotechnology [11-13]. It relies on an exposed Watson-Crick sticky region (i.e., sticky single-stranded end) on DNA duplex, which is used to anchor the invading strand, causing displacement with one of the strands in the DNA duplex [14]. Toehold reactions have significantly increased the reaction rate of DNA strand displacement, and greatly facilitate the development of DNA nanotechnology [15]. Various DNA nanodevices have been constructed, such as in the field of DNA machines, Fang et al. reported a three-dimensional lame DNA walker [16]; in the field of DNA computing, Winfree et al. used seesaw reactions to construct DNA logic gate components [17]; in the field of biosensing, a biosensor for detecting miRNA at the attomolar level was constructed using strand displacement reactions [18]. Meanwhile, many researchers are committed to developing regulatory tools for toehold-mediated DNA strand displacement reactions, enabling DNA nanodevices to achieve more complex functionality such as precise control of reaction rate and reusability of reaction systems [19-21].
DNA nanotechnology based on toehold reactions has achieved significant progress and widespread applications comprehensively. However, it is also subject to a fundamental restriction, which is the requirement of a sticky end to initiate the reaction [22]. It is known that a sticky end means an unstable activated state prone to promulgate many undesired side reactions [23,24]. Therefore, the leakage problem and huge endeavor of leakage prevention have been the longstanding focus since the birth of the technology [25]. Basically, researchers need to use carefully designed DNA strands and the three-letter principle to inhibit potential side reactions [17]. However, as the reaction scale increases, it becomes difficult to suppress leakage with only sequence design of the main reaction. Later on, some researchers proposed leakage inhibition methods that do not rely on sequence design. For example, Song et al. used Shadow Cancellation to restrict leakage in the main circuit [26]. Nonetheless, this strategy has a very complex secondary structure and requires additional nucleic acid sequences beyond the main reaction sequence, increasing the complexity of sequence design. Hu et al. proposed the method using a high-binding DNA-locked nucleic acid (LNA) bond to inhibit the breathing effect [27], but modifying locked nucleic acid on DNA is also challenging. Overall, the entire DNA nanotechnology is currently built on toehold-mediated DNA strand displacement, in which the artificial toehold region causes an intrinsic tendency of leakage. We believe that searching for a toeless blunt-end DNA strand displacement reaction pattern would be a promising attempt and will enrich the basic toolbox of DNA nanotechnology.
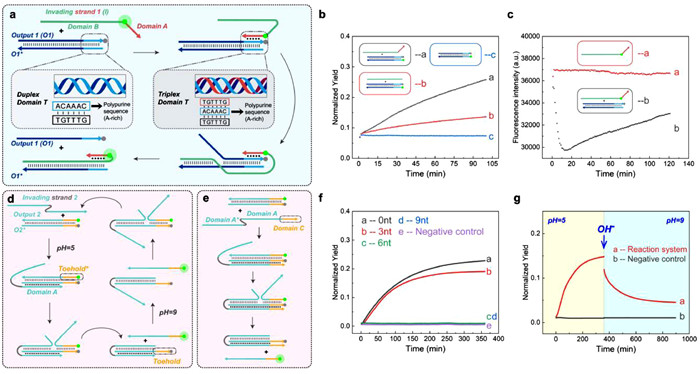
Herein, we developed a toeless and reversible DNA strand displacement reaction based on Hoogsteen-bond. As shown in Fig. 1a, the Invading strand 1 (denoted as I strand) consists of Domain A and Domain B. Domain A is designed to be T-rich and Domain B is complementary to the O1* strand of the target dsDNA. At the same time, the duplex domain T of Output 1 (O1) strand is designed to be A-rich. Therefore, the I strand's T-rich Domain A would form Hoogsteen-bonds with the A-rich Domain T of the double-stranded DNA (dsDNA) [28]. The interesting point is that those Hoogsteen-bonds could serve as a trigger to lead the Domain B further displacing off the O strand, and the final product would be I strand/O* strand hybrid with the Domain T being triplex. As the formation of Hoogsteen-bond involves the invasion of T-rich single strand on A-rich du-plex strand, the trigger we constructed for driving strand displacement reaction will not contain a single-stranded toehold, thus eliminating the leakage introduced by sticky ends at the source. The reaction in Fig. 1a is denoted as reaction mode 1. In this reaction mode, triplex-forming oligonucleotide binds to the polypurine oligonucleotide strand in an antiparallel orientation (denoted as antiparallel Hoogsteen-bond, aPHB). Moreover, if we design the triplex-forming oligonucleotide in parallel orientation to the polypurine strand of DNA (denoted as parallel Hoogsteen-bond, PHB), then when we adjust the pH from a relatively low level of 5 to a higher level of 9, the protonated cytosines in triplex domain will undergo deprotonation [29,30]. As a result, the PHB will be broken, and the strand displacement reaction could be possibly reversed. We will validate the above principle through experiments in the follow-up study and further adjust the reaction strategy to accelerate the reaction rate and enhance the reaction degree. Eventually, we will use this principle to construct two types of DNA nanomachines: DNA logic gate components and DNA walker. We believe that our established toeless reversible strand displacement would be widely applied in various DNA nanodevices.
Figure 1
Figure 1.
(a) A Schematic illustration of the principle of triplex-mediated DNA strand displacement reaction mode 1. (b, c) The influence of the triplex invasion domain of the invading strand on strand displacement reaction mode 1, with the control group consisting of 1 µmol/L Invading strand 1 (labeled with FAM). (d, e) A Schematic illustration of the basic principle of reaction mode 2. (f) The influence of different lengths of Domain C of Invading strand 2 on the reaction degree of reaction mode 2. The negative control group consisted of 1 µmol/L Output 2/O2* duplex complex (labeled with FAM/BHQ1, respectively), and the positive control group consisted of 1 µmol/L Output 2 (labeled with FAM). (g) The validation process of reaction system resetting under reaction mode 2 (the experimental group, negative control group, and positive control group used Output 2 or O2* labeled with Cy5 or BHQ2, respectively).
Firstly, we validated the basic principle of the reaction. We labeled the quencher BHQ1 at the end of Output 1 (O1) (denoted as O1-BHQ1), and the fluorophore FAM at the end of O1*. We hybridized O1 and O1* at a concentration of 1 µmol/L as the reaction substrate. We added 1 µmol/L unlabeled I strand to group 1, and added simulated leaky strand 2 without the triplex binding region to group 2. Both groups received 1 µmol/L of reaction substrate at the same time. In addition, we set 1 µmol/L reaction substrate as the negative control group and 1 µmol/L O1* strand as the positive control group. Subsequently, we conducted the reactions for 100 min at pH 7 and 37 ℃ for each group, while detecting the fluorescence intensity of each group at 1-min intervals (with the positive control group set as the reference value of 1 or 100% in all experiments). Fig. 1b indicates that, the reaction rate of group 1 is significantly faster, compared to that of group 2, suggesting that Domain A can mediate the toeless DNA displacement reaction. Next, to confirm that the reaction mechanism of toeless DNA displacement reaction is indeed as shown in Fig. 1a, we labeled fluorophore FAM at the boundary of Domain A and Domain B of Invading strand 1 (denoted as Invading strand 1-FAM). In the experiment, we added 1 µmol/L Invading strand 1-FAM to the prehybridized substrate of 1 µmol/L O1-BHQ1 and O* as the experimental group, and 1 µmol/L Invading strand 1-FAM was set as the control group. Subsequently, we conducted the reactions for 120 min at pH 7 and 37 ℃ for each group, while detecting the fluorescence intensity of each group at 1-min intervals. As shown in Fig. 1c, compared with the control group, the fluorescence intensity of the experimental group showed a significant decrease followed by an increase, indicating that the rapid binding of Domain A and polypurine sequence mediated the subsequent strand displacement reaction. We believe that the length of Domain A is a key factor affecting the kinetics and thermodynamics of toeless triplex-mediated DNA displacement reaction. Increasing the length of Domain A not only enhances the binding strength between Domain A and Domain T, but also increases the difficulty of dissociation of Output 1 strand. Therefore, theoretically, there is an equilibrium point for the dissociation and binding tendency, which corresponds to a moderate value of the length of Domain A. Deviation from the optimal length in Domain A, either longer or shorter, leads to a decrease in reaction efficiency. Therefore, we further vary the length of Domain A and Domain T to explore the effect of triplex domain length on the degree of reaction. According to the results in Fig. S1 (Supporting information), this equilibrium is reached when the length of triplex domain is 9 nucleotides. In conclusion, we have successfully constructed a toeless triplex-mediated DNA strand displacement reaction with aPHB.
Next, we attempted to construct reaction mode 2 using PHB. And based on reaction mode 2, we further constructed a pH-responsive and reversible reaction. As shown in Figs. 1d and e, at pH around 5, Invading strand 2 forms PHB with O2* and displaces Output 2. We designed a dissociation domain Toehold* at the end of Output 2, and the complementary domain is denoted as Toehold. When the pH increases to around 9, the PHB in the triplex domain dissociates. At this point, the Toehold can mediate the re-hybridizing of Output 2 with O2* and displace Invading strand 2, thus completing the reverse of the reaction. Before conducting the reversible reaction study, we aimed to investigate the influence of the length of Domain C, which forms the triplex structure, on the reaction kinetics and thermodynamics (Fig. 1e). 1 µmol/L Invading strand 2 of different Domain C lengths is used to react with 1 µmol/L of Output 2-O2* duplex complex as experimental groups. We also set a negative control group of 1 µmol/L Output 2-O2* duplex complex and a positive control group of 1 µmol/L Output 2 only. Subsequently, we conducted the reactions for 360 min at pH 7 and 37 ℃ for each group, while detecting the fluorescence intensity of each group at 1-minute intervals. We discovered that the reaction could only occur when the length of Domain C was less than 6 nt (Fig. 1f). Moreover, the reaction was most efficient when the length of Domain C was reduced to 0 nt. We further studied the reactions when the length of Domain C was 6, 5, and 4 nt (Fig. S2 in Supporting information), which confirmed that the length of Domain C required for the reaction to occur was indeed at least 6 nt. Similar to reaction mode 1, we also explored the effect of reducing the length of Domain A on the efficiency of the reaction. When the length of Domain C was fixed at 0 nt and the dissociation region length was constant, we found that the efficiency of reaction mode 2 rose as the length of Domain A decreased (Fig. S3 in Supporting information). Through these two experiments, we demonstrated that the difficulty of output strand dissociation from the original triplex domain significantly impacted on the efficiency of the reaction mode 2, which provided feasibility for accurate manipulation of this reaction. Based on the above experiments, we explored the reversibility of this reaction mode by adjusting the pH. As shown in Fig. 1g, we adjusted the pH of the system from 5 to 9 and continued to detect fluorescence at 1-min intervals, 37 ℃. We observed a rapid decrease in fluorescence signal, indicating that the reverse reaction occurred and the reaction system was reverse. This result validated the feasibility of the reaction strategy shown in Fig. 1d. In summary, we successfully constructed a reversible toeless strand displacement reaction using PHB.
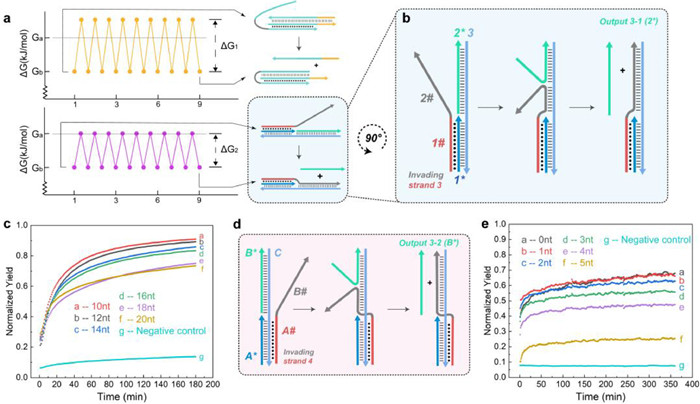
The above two reaction modes have achieved toeless blunt-end DNA strand displacement reaction. However, their reaction kinetics are not ideal, and the reaction degree is generally less than 30%, which limits their further application and expansion. Hoogsteen-bond must rely on the Waston-Crick bond to exist stably [28]. Thus, both of the aforementioned toeless triplex-mediated strand displacement reactions involve a repeated process of Hoogsteen-bond dissociation and formation, which greatly increases the energy barrier of each strand displacement step and leads to a decrease in the reaction rate (Fig. 2a). Therefore, through further optimization, we designed the following two reaction modes to solve the above problems. First, the principle of reaction mode 3 is shown in Fig. 2b. After the 1# domain of Invading strand 3 forms a PHB with the 1* strand, the 2# domain will replace the Output 3–1 (2*) strand and bind to the 3 strand. At the same time, the 2* strand will react with the reporter system labeled by FAM fluorophore and BHQ1 quencher, generating a fluorescence signal. This reaction does not involve the repeated process of Hoogsteen-bond formation and dissociation. The energy barriers that need to be overcome for each base substitution step in this strand displacement reaction are lower. Thus, theoretically, the efficiency of reaction mode 3 is higher than modes 1 and 2, and the reaction rate is also faster. At the same time, the strength of PHB is related to the degree of protonation of the pyrimidine in strand 1*. Hence, by gradient adjusting the length of the triplex domain in the strand displacement reaction and the dissociation region length in the strand migration reaction, we can achieve precise control over the kinetics and thermodynamics of the reaction. We added 1 µmol/L of Invading strand 3, which have different lengths of 10–20 nt in Domain 1, to the preformed 1*−2*−3* duplex complex with a concentration of 1 µmol/L, and added 1 µmol/L of reporter system to each group as the experimental group. The negative control group consisted of 1 µmol/L 1*−2*−3* complex without Invading strand 3 and 1 µmol/L reporter system, and the positive control group consisted of only 1 µmol/L Output 3–1 and 1 µmol/L reporter system. Subsequently, we allowed each group to react for 180 min at pH 5 and 37 ℃, and detected the FAM fluorescence intensity of each group every 1 min. The results in Fig. 2c show that the reaction efficiency of reaction mode 3 is generally between 70%–90%, and can be completely reacted within 2 h. Compared with reaction modes 1 and 2, reaction mode 3 does have a higher reaction efficiency and a faster reaction rate. At the same time, we observed that as the length of Domain 1 in strand 3 increased, the efficiency of the triplex strand displacement reaction decreased. We speculate that this is due to a significant increase in the invading reaction energy barrier with an increase in the length of the triplex domain. This result reveals that an increase in the length of the triplex domain significantly increases the binding energy barrier between the invading strand and the reaction substrate. In subsequent similar strand displacement experiments, the length of the triplex region to 10 nt will be set by default.
Figure 2
Figure 2.
(a) Differences in energy barriers between reaction mode 2 and reaction mode 3. (b) A Schematic illustration of the basic principle of reaction mode 3. (c) The relationship between the length of the triplex domain and the reaction degree of reaction mode 3. The negative control group consisted of 1 µmol/L 1*−2*−3* complex and 1 µmol/L reporter system, and the positive control group consisted of 1 µmol/L Output 3–1 and 1 µmol/L reporter system (reporter system labeled with pair of FAM/BHQ1). (d) A Schematic illustration of the basic principle of reaction mode 4. (e) The relationship between the length of the dissociation region and the reaction degree of reaction mode 4. The negative control group consisted of 1 µmol/L A*-B*-C* complex and 1 µmol/L reporter system, and the positive control group consisted of 1 µmol/L Output 3–2 and 1 µmol/L reporter system (reporter system labeled with FAM/BHQ1, respectively).
As shown in Fig. 2d, based on the aPHB not relying on protonated cytosine, we designed reaction mode 4 that is not influenced by pH changes. The A# domain of Invading strand 4 formed aPHB with A* and C strands, while its B# domain displaced the Output 3–2(B*) strand. The resulting B* strand reacted with the reporter system containing FAM-BHQ1 pair to generate fluorescence (related reporter system sequences shown in Supporting information 109 to 110). In addition to Invading strand 4 that can undergo complete strand displacement reaction with B*, we also designed Invading strands 4 with decreasing length at the 3′ end. These strands can create a dissociation region at the end of the A*-B*-C* complex, for observing the length effect of the dissociation region on the reaction degree. We shortened the B# 3′ ends by 1 nt for each chain, denoted as Invading strand 4–0 nt to Invading strand 4–5 nt (related sequences shown in Supporting information 10–3 to 10–8). Each of the Invading strand 4-n (n = 0–5 nt) strands was added to the 1 µmol/L A*-B*-C duplex complex at a concentration of 1 µmol/L as the experimental group. The negative control group was the 1 µmol/L A*-B*-C* complex without Invading strand 4-n and the reporter system at a concentration of 1 µmol/L, while the positive control group was only the 1 µmol/L reporter system with 1 µmol/L Output 3–2. Each group was allowed to react for 360 min at pH 7 and 37 ℃, and the FAM fluorescence intensity was detected at 1-min intervals. As shown in Fig. 2e, the reaction degree of reaction mode 4 gradually decreased as the length of the dissociation region reserved at the B# 3′ end increased. The highest reaction degree, which reached about 65%, was achieved when the length of the dissociation region was 0 nt, suggesting a complete chain displacement reaction in the system. Under reaction mode 4, the triplex structure has two binding patterns: T-A:T and A-A:T. We found that the rate of strand displacement reaction with T-A:T binding was slightly faster than that with A-A:T binding, as the result shown in Fig. S4 (Supporting information). Yet, when the length of the triplex domain and the dissociation region were kept constant, the final reaction degree was close to 65% regardless of whether we mixed adenine (A) or thymine (T) bases into the invading region of Invading strand 4. Therefore, we conclude that the principle of reaction mode 4 has been validated, indicating that our toeless triplex-mediated strand displacement reaction can occur independently of pH changes.
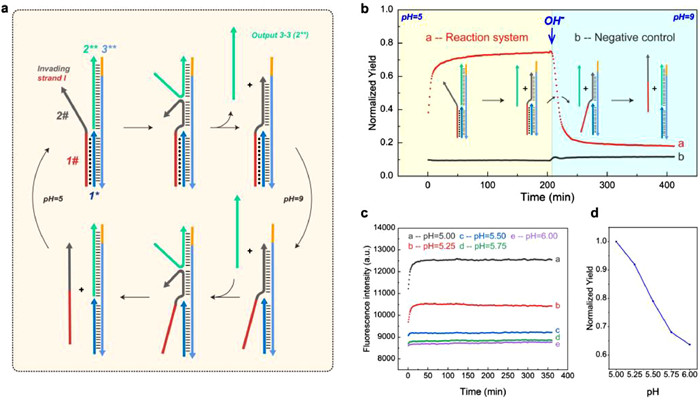
After verifying the reaction mechanisms of the aforementioned two reaction modes, we explored the reversibility of reaction mode 3. As seen in Fig. 3a, we added a 6-nt dissociation region to the 3 strand of reaction mode 3 (denoted the new strand as 3**) which can be used as a toehold-mediated strand displacement reaction in the reverse process. In addition, we also modified the reporter pair into a Cy5-BHQ2 combination to limit the effect of pH changes on the fluorescence of the reaction. We conducted a reaction at pH 5 and 37 ℃ using 1 µmol/L of Invading strand I and 1 µmol/L of the 1*−2**−3 duplex complex, which eventually showed a similar forward reaction degree as reaction mode 3. The negative control group consisted of a 1 µmol/L 1–2*−3** complex without the addition of Invading strand I and a 1 µmol/L reporter system, whereas the positive control group consisted of only 1 µmol/L Output 3–3 and 1 µmol/L reporter system. After adjusting the pH of the system from 5 to 9, we immediately recorded the fluorescence intensity of each group at 1-min intervals at 37 ℃. As shown in Fig. 3b, there is a rapid decrease in the fluorescence signal in the system from the platform value, indicating the reversible nature of reaction mode 3. Furthermore, we evaluated the correlation between reaction mode 3′s reaction thermodynamics and pH. As illustrated in Figs. 3c and d, we observed that the degree of the above forward reaction decreased with increasing pH in the range of pH 5–6. This demonstrates that we can precisely regulate the reaction degree of reaction mode 3 by adjusting pH. Additionally, based on this principle, we demonstrated that the output strand of toeless triplex-mediated strand displacement reaction can serve as input for the same reaction, i.e., this type of strand displacement has the potential for modular self-compatibility, enabling the construction of cascade reactions (combing modes 3 and 4). The sequence design and reaction results are shown in Figs. S5a–c. We also investigated the impact of reaction temperature on our toeless triplex-mediated strand displacement reaction. The results are shown in Fig. S6 (Supporting information). Based on the design of cascade reaction DNA sequences, the final reaction degree of the cascade reaction was used as the dependent variable, while the reaction temperature was varied from 31 ℃ to 46 ℃. We observed no significant differences in the final reaction degree when the reaction temperature was at or below 37 ℃. However, the reaction rate was notably faster at 37 ℃ (the local ambient temperature being 30 ℃, which is commonly encountered during the transfer of experimental samples). As the temperature increased further, both the reaction degree and rate significantly decreased. We infer that when the temperature increases, the binding degree of all DNA duplex components in the system decreases, and the high temperature also makes it difficult to form Hoogsteen-bond [28]. This finding indicates that our reaction operates optimally at a temperature around 37 ℃, consistent with normal human body temperature. This provides a foundation for integrating our reaction into in vivo biological circuits.
Figure 3
Figure 3.
(a) A Schematic illustration of the fundamental principle of resetting reaction mode 3, involving the addition of a dissociation region at the end of the 3 strand. (b) The verification process for resetting the reaction mode 3. The negative control group consisted of a 1 µmol/L 1*−2**−3** complex and a 1 µmol/L reporter system, while the positive control group consisted of 1 µmol/L Output 3–3 and 1 µmol/L reporter system (reporter system labeled with Cy5/BHQ2, respectively). (c, d) The relationship between small-range pH adjustment and variations in reaction degree within the reaction mode 3 system.
In summary, the toeless triplex-mediated strand displacement reaction modes that we have invented completely circumvents the leakage problem of the traditional single-strand base-exposed sequence in W-C sticky end toehold mediated strand displacement reaction, and incorporates both reversible and regulatory capabilities. Next, we shall illustrate its strong compatibility in DNA logic circuits.
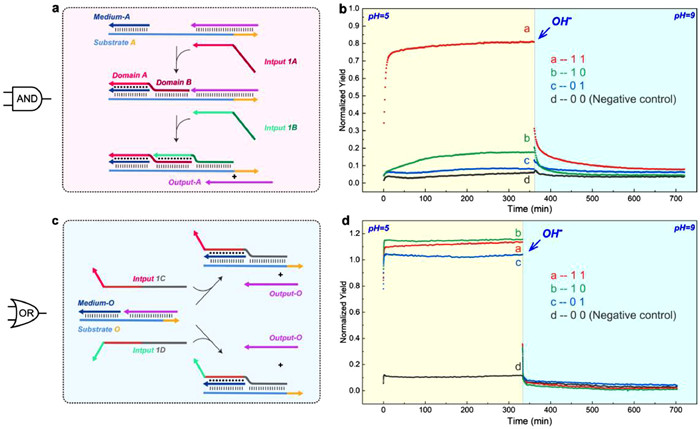
Based on reaction mode 3, we aimed to explore the possibility of constructing simple DNA circuit components, AND gate and OR gate. As shown in Fig. 4a, the AND gate consists of Substrate A, Medium-A, Output-A, and two types of Input 1 strands. When Domain A on the Input 1A strand binds with the established substrate complex, Domain B in the migration region serves as a mediator, facilitating Input 1B strand to form Hoogsteen-bonds with Domain B. Therefore, only when both attached Input 1A and Input 1B are present in the system, can Domain A mediate the invading of the Input 1B strand and replace the Output-A strand. Subsequently, the Output-A strand and Cy5-BHQ2 reporter system react to produce a Cy5 fluorescence signal. We reserved a dissociation region at the end of the Substrate A strand. When the system pH was adjusted to 9, all triplex region will be dissociated. At this point, Input A falls off naturally, and Output-A can replace Input 1B strand for the reaction to be reversed. We employed the Substrate A/Medium-A/Output-A duplex complex as the reaction substrate. In the first group, 1 µmol/L Input 1A and 1 µmol/L Input 1B were introduced to the 1 µmol/L substrate as signal (1 1). In group 2 (1 0) and group 3 (0 1), we introduced either 1 µmol/L Input 1A or 1 µmol/L Input 1B respectively to the same 1 µmol/L substrate. The reporter system was added at a concentration of 1 µmol/L in each experimental group. We used the 1 µmol/L substrate complex without any input and 1 µmol/L reporter as negative controls (0 0), and the system only with 1 µmol/L Output-A and 1 µmol/L reporter as positive controls. We adjusted the pH to 5 in each group, and recorded the fluorescence intensity every 1 min at 37 ℃ (recorded every 5 s within 6 min after each addition). As shown in Fig. 4b, we observed a rapid and significant fluorescence signal in the (1 1) reaction group, while each individual Input 1A or 1B alone did not generate a significant signal. In addition, there was a rapid reverse of the reaction system after pH adjustment to 9. These results demonstrate the successful construction of our reversible toeless triplex-mediated AND gate.
Figure 4
Figure 4.
(a, b) A Schematic illustration and relevant experimental validation of an AND gate based on a triplex-mediated strand displacement reaction. The negative control group consisted of a 1 µmol/L Substrate A/Medium-A/Output-A duplex complex with a 1 µmol/L reporter system, while the positive control group consisted of a 1 µmol/L Output-A and 1 µmol/L reporter system (reporter system labeled with Cy5/BHQ2, respectively). (c, d) A Schematic illustration and relevant experimental validation of an OR gate based on a triplex-mediated strand displacement reaction. The negative control group consisted of a 1 µmol/L Substrate O/Medium-O/Output-O duplex complex with a 1 µmol/L reporter system, while the positive control group consisted of a 1 µmol/L Output-O and 1 µmol/L reporter system (reporter system labeled with Cy5/BHQ2, respectively).
The reaction mechanism of the OR gate is illustrated in Fig. 4c. We used two partially identical sequences, Inputs 1C and 1D, to represent two input signals 1. Both Input signals can trigger the same strand displacement reaction and lead to the same Output-O output. We defined Output-O as responding to the Cy5-BHQ2 reporter system and generating Cy5 fluorescence as output signal 1. This logic gate can also be reversed by the toehold domain reserved on the substrate O strand when the reaction system is raised to pH 9. We combined Substrate O/Medium-O/Output-O duplex complex as reaction substrate. In group 1 (1 1), 1 µmol/L substrate, 1 µmol/L Input 1C, and 1 µmol/L Input 1D were added. In group 2 (1 0) and group 3 (0 1), we added 1 µmol/L substrate and 1 µmol/L of either Input 1C or 1 µmol/L Input 1D, respectively. Moreover, we added 1 µmol/L reaction substrate and 1 µmol/L reporter system to the negative control group (0 0). At the same time, we added 1 µmol/L Output-O and 1 µmol/L reporter system to the positive control group. Finally, we measured fluorescence intensity every minute at pH 5 and 37 ℃ for each group (recorded every 5 s within 6 min after each addition). The results shown in Fig. 4d demonstrated that all experimental groups exhibited significant fluorescence signals, while the negative control group showed almost no signal. Once the pH was adjusted to 9, all reaction systems were reversed. This result proved that we have successfully constructed a reversible toeless triplex-mediated OR gate.
The results of the above AND and OR gates demonstrate that modular and reversible DNA circuit logic gate components can be constructed based on the toeless triplex-mediated DNA displacement mechanisms utilized in this study. This reverse does not generate any type of waste, and thus does not adversely affect subsequent computations.
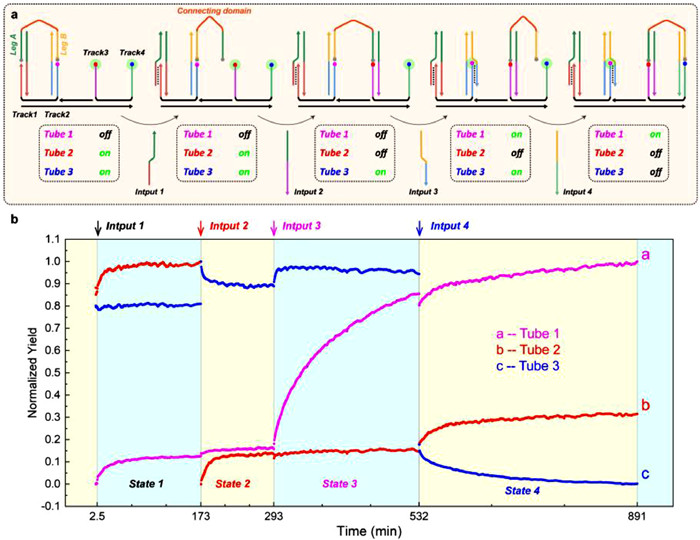
The validation of the aforementioned reaction concept leads us to believe that our toeless triplex-mediated strand displacement reaction can undertake more complex tasks. Consequently, based on the toeless triplex-mediated strand displacement mechanism, we designed a DNA nano walking machine, namely DNA Walker. The operating concept of this Walker integrates reaction modes 3 and 4 (In PHB and aPHB mechanism, the orientation of invading domain is opposite). We accomplished the alternating movement of this walking structure by adjusting the design of input and the sticky end sequences of track strand. Fig. 5a demonstrates that the walking strand is composed of Leg A, Leg B, and the connecting domain. We have labeled the quencher BHQ1 at both ends of Leg A and Leg B. The substrate Track complex has four free sticky single-strand ends, Track1 to Track4, with FAM fluorophore labeling on the free sticky single-strand ends of Track 2 to Track 4. Prior to the start of the reaction, we added the walking strand into the previously assembled Track complex as the initializing state. In State 1, Input 1 was introduced into Track complex to induce detachment of Leg A. In State 2, Leg A was hybridized with Track 3 resulting in quenching of the fluorophore at the end of Track 3. In State 3, Input 3 was added to unquench the fluorophore on Track 2. Subsequently, in State 4, Leg B was hybridized with Track 4 through the addition of Input 4 followed by quenching of the fluorophore on the Track 4. These four states constitute a complete walking cycle: Leg A and Leg B initially bind to Track 1 and Track 2, respectively, and ultimately move to Track 3 and Track 4, respectively. Given that the conformational changes of the Track complex may cause interference between FAM fluorescence signals (Fig. S7 in Supporting information), we set up three independent tubes. The Track complex structures in each tube are the same, but the positions of the FAM fluorophore located correspond to three different endpoints of the steps. We marked the observation of fluorescence at a certain position as "on" and the disappearance of fluorescence as "off". In the above reactions, the concentrations of each reactant were all 1 µmol/L, and the reaction conditions were pH 5 and 37 ℃. The results in Fig. 5b showed that after adding of inputs at each step, the fluorescence signals of all three tubes exhibited the expected changes, which shows that each reaction proceeded as expected, and the walking cycle was finally completed. Additionally, the fast reactions between State 1/State 2 and State 3/State 4, as well as the relatively slow increase in fluorescence between State 2/State 3. This further confirmed that duplex hybridization, toeless triplex-mediated strand displacement, and another duplex hybridization occurred in the three State sequentially. The results suggest that an active DNA nanomachine can be constructed based on the reaction modes of our experiment. Theoretically, various cargoes can be connected to the connecting domain to form a cargo complex through covalent or Watson-Crick bonding modification. By designing the sequences of the sticky regions of Input No.n and Track No.n in each step, specific hybridization can be achieved, thereby controlling the stable transportation of the cargo complex on a Track complex of arbitrary length. The construction of the aforementioned logic gates and walker demonstrates that our reaction strategy can be flexibly applied to various DNA nanomachine, with high modularity and highly adjustable reaction processes, and has a great application potential.
Figure 5
Figure 5.
(a) A schematic illustration and relevant experimental validation of the DNA walker principle. Leg A/Leg B step from Track 1/Track 2 to Track 3/Track 4, respectively. In this context, the fluorophore of the Track complex in Tube 1 was labeled at the end of Track 2, while the fluorophore of the Track complex in Tube 2 was labeled at the end of Track 3, and the fluorophore of the Track complex in Tube 3 was labeled at the end of Track 4. (b) In each of the 4 states of in walking cycle, 3 tubes exhibit different fluorescence changes, corresponding to 3 types of walking structure with different locations of fluorophore.
At last, as a summary of the full study, we have designed a novel DNA strand displacement reaction that enables the regulation of reaction degree and direction dependent on pH changes between 5 and 9, while avoiding various drawbacks of exposed sticky single-strand ends. We discovered that the proposed reaction can be finely regulated within the pH range of 5–6, and this pH range is very prevalent in biological organisms. Additionally, with the improvement of the reaction process, the degree of the reaction can reach up to 90%, which is close to the efficiency of traditional toehold-mediated strand displacement reactions, indicating that our toeless reaction still maintains the advantage of reaction efficiency in toehold reaction. Moreover, we verified our reaction can be integrated into DNA logic gates, which are also pH-responsive and not affected by leakage interference from Watson-Crick base pairing, and can be used as the trigger or regulatory components of complex circuits that require pH changes as switches. The successful validation of the DNA Walker mechanism for 4 steps cycle indicates that our reaction modes are capable of constructing more complicated DNA molecular machines. We expect that the toeless triplex-mediated strand displacement response will be widely used in the development of DNA circuits, molecular sensors, and other complex biological dynamic systems.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Yang Qin and Jiangtian Li contributed equally to this work and should be considered co-first authors. All authors have given approval to the final version of the manuscript.
This work was financially supported by the National Key Research and Development Program of China (No. 2021YFC2701402), the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University (No. Sklb2021-k06), the Open Foundation of NHC Key Laboratory of Birth Defect for Research and Prevention (Hunan Provincial Maternal and Child Health Care Hospital) (No. KF2020007), and the Open Foundation of Translational Medicine National Science and Technology Infrastructure (Shanghai) (No. TMSK-2021–141).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108826.
A. Idili, A. Vallee-Belisle, F. Ricci, J. Am. Chem. Soc. 136 (2014) 5836–5839. doi: 10.1021/ja500619w
[30]
A. Amodio, A.F. Adedeji, M. Castronovo, E. Franco, F. Ricci, J. Am. Chem. Soc. 138 (2016) 12735–12738. doi: 10.1021/jacs.6b07676
Figure 1
(a) A Schematic illustration of the principle of triplex-mediated DNA strand displacement reaction mode 1. (b, c) The influence of the triplex invasion domain of the invading strand on strand displacement reaction mode 1, with the control group consisting of 1 µmol/L Invading strand 1 (labeled with FAM). (d, e) A Schematic illustration of the basic principle of reaction mode 2. (f) The influence of different lengths of Domain C of Invading strand 2 on the reaction degree of reaction mode 2. The negative control group consisted of 1 µmol/L Output 2/O2* duplex complex (labeled with FAM/BHQ1, respectively), and the positive control group consisted of 1 µmol/L Output 2 (labeled with FAM). (g) The validation process of reaction system resetting under reaction mode 2 (the experimental group, negative control group, and positive control group used Output 2 or O2* labeled with Cy5 or BHQ2, respectively).
Figure 2
(a) Differences in energy barriers between reaction mode 2 and reaction mode 3. (b) A Schematic illustration of the basic principle of reaction mode 3. (c) The relationship between the length of the triplex domain and the reaction degree of reaction mode 3. The negative control group consisted of 1 µmol/L 1*−2*−3* complex and 1 µmol/L reporter system, and the positive control group consisted of 1 µmol/L Output 3–1 and 1 µmol/L reporter system (reporter system labeled with pair of FAM/BHQ1). (d) A Schematic illustration of the basic principle of reaction mode 4. (e) The relationship between the length of the dissociation region and the reaction degree of reaction mode 4. The negative control group consisted of 1 µmol/L A*-B*-C* complex and 1 µmol/L reporter system, and the positive control group consisted of 1 µmol/L Output 3–2 and 1 µmol/L reporter system (reporter system labeled with FAM/BHQ1, respectively).
Figure 3
(a) A Schematic illustration of the fundamental principle of resetting reaction mode 3, involving the addition of a dissociation region at the end of the 3 strand. (b) The verification process for resetting the reaction mode 3. The negative control group consisted of a 1 µmol/L 1*−2**−3** complex and a 1 µmol/L reporter system, while the positive control group consisted of 1 µmol/L Output 3–3 and 1 µmol/L reporter system (reporter system labeled with Cy5/BHQ2, respectively). (c, d) The relationship between small-range pH adjustment and variations in reaction degree within the reaction mode 3 system.
Figure 4
(a, b) A Schematic illustration and relevant experimental validation of an AND gate based on a triplex-mediated strand displacement reaction. The negative control group consisted of a 1 µmol/L Substrate A/Medium-A/Output-A duplex complex with a 1 µmol/L reporter system, while the positive control group consisted of a 1 µmol/L Output-A and 1 µmol/L reporter system (reporter system labeled with Cy5/BHQ2, respectively). (c, d) A Schematic illustration and relevant experimental validation of an OR gate based on a triplex-mediated strand displacement reaction. The negative control group consisted of a 1 µmol/L Substrate O/Medium-O/Output-O duplex complex with a 1 µmol/L reporter system, while the positive control group consisted of a 1 µmol/L Output-O and 1 µmol/L reporter system (reporter system labeled with Cy5/BHQ2, respectively).
Figure 5
(a) A schematic illustration and relevant experimental validation of the DNA walker principle. Leg A/Leg B step from Track 1/Track 2 to Track 3/Track 4, respectively. In this context, the fluorophore of the Track complex in Tube 1 was labeled at the end of Track 2, while the fluorophore of the Track complex in Tube 2 was labeled at the end of Track 3, and the fluorophore of the Track complex in Tube 3 was labeled at the end of Track 4. (b) In each of the 4 states of in walking cycle, 3 tubes exhibit different fluorescence changes, corresponding to 3 types of walking structure with different locations of fluorophore.

 DownLoad:
DownLoad:

 下载:
下载:






