Key Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, Shanghai Key Laboratory of Functional Materials Chemistry, East China University of Science and Technology, Shanghai 200237, China
b.
Yaopu (Shanghai) Pharmaceutical Technology Co., Ltd., Shanghai 201108, China
Received Date:
17 December 2022 Accepted Date:
29 May 2023 Revised Date:
27 May 2023 Available Online:
15 December 2023
Abstract:
The mitigation of under-coordinated Pb2+ (halide vacancy) defect remains an imperative challenge in the perovskite solar cells, especially printable mesoscopic perovskite solar cells (FP-PSCs). Here we report a commercial-available polyazin anticancer drug Sapanisertib as coordination passivator of halide vacancies in FP-PSCs, thereby achieving the photoelectric conversion efficiency (PCE) to 18.46%, along with a record certified PCE of 18.27%. In polazin Sapanisertib (Sap), there exists two kinds of nitrogen atoms: in-aromatic ring (in purine and oxazole rings, IAR-Ns) and out-aromatic ring (substituted amino groups, OAR-Ns). Through multiple characterizations, and DFT calculations show that substituted amino groups OAR-Ns hardly get interaction with the halide vacancy due to the distribution of charge density in Sapanisertib. Our work suggests that the selective coordination is of great significance for the design of high-performance passivators for printable mesoscopic perovskite solar cells.
For organic-inorganic lead halide perovskite, the conduction band is composed of sigma antibonding orbitals Pb-6p and I-5p, and the valence band consists from deep to shallow of Pb-6s and I-5p sigma bonding orbitals, Pb-6p and I-5p sigma bonding orbitals, and Pb-6s and I-5p sigma antibonding orbitals, respectively [1,2]. Molecular dynamics simulations revealed that the Pb defects due to iodine migration and vacancies lead to the formation of Pb-Pb bonds in close proximity to their neighboring Pb atoms and the formation of deep defective energy levels, which in turn cause the Pb-I bonds near the two Pb atoms to further collapse and form new shallow defective energy levels [3]. The existence of halide vacancies would introduce nonradiative Shockley-Read-Hall recombination limiting the photovoltaic performances of perovskite solar cells. Reducing the under-coordinated Pb2+ (halide vacancy) defects is expected as a key pathway to improve the carrier injection efficiency with passivators [4–8].
Organic passivators are focused on herteroatomic derivatives since such N, O, S and P are easy to adsorb with Pb2+ defects via lone pairs [9–20]. For example, addition of 3-methyl-2,4-nonanedione to chlorobenzene antisolvent can combine with insufficiently coordinated Pb2+ ions in CH3NH3PbI3 perovskite films, thereby increasing the open-circuit voltage (Voc) of PSCs by 52 mV [21–24]. The carbonyl groups in 2,2-difluoropropanediamide (DFPDA) can form chemical bonds with under-coordinated Pb2+ defects, reducing perovskite crystallization rate and improving its film quality. Similary, the amino groups not only inhibit ion migration of iodide but also increase the passivation effect of carbonyl groups through changing electron density [25–27]. Two conjugated carbonyl groups in 1,3,7-trimethylxanthine (caffeine) can coexist with four nitrogen atoms. As expected, the strong interaction between carbonyl group and under-coordinated Pb2+ can increase the activation energy during film crystallization [28,29].
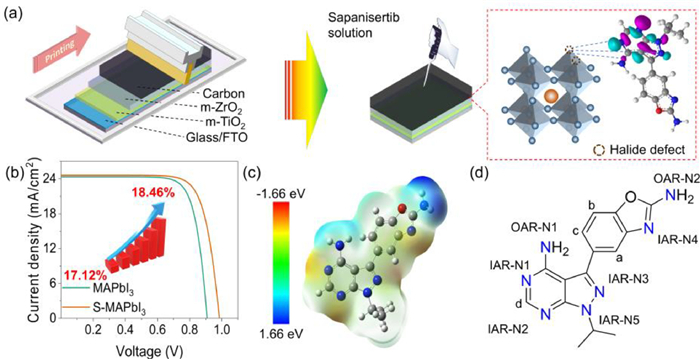
Fully-printable mesoscopic perovskite solar cells (FP-PSCs) have been widely concerned since proposed by Han et al. in 2014. The desirable benefit with printing technology is that can be assembled in air and long-term stability (Fig. 1a) [30]. However, when compared with conventional devices, FP-PSCs often suffer from more serious halide vacancies and lower carrier transport efficiency due to spontaneous liquid diffusion and solvent evaporation process [31]. So far, the highest certified power conversion efficiency(PCE) has reached only 17.7% in FP-PSCs [32]. In order to further inhibit Shockley-Read-Hall recombination in FP-PSCs, here we report a polyazin anticancer drug Sapanisertib to coordinate with under-coordinated Pb2+ (halide vacancy), behaving as the selective coordination for high-performance passivators.
Figure 1
Figure 1.
Structural information of the Sapanisertib molecule and its action process in FP-PSCs. (a) Schematic illustrations of IAR-N passivation for under-coordinated Pb2+ in perovskites over OAR-N in the Sapanisertib molecular environment. (b) J-V curve of MAPbI3 devices and S-MAPbI3 devices. (c) Molecular surface electrostatic potential of Sapanisertib. (d) Chemical structure and different kinds of heteroatoms of Sapanisertib.
Using several characterization methods, such as structural splitting, Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) calculations, we took insight into the under-coordinated Pb2+ (halide vacancy) with two kinds of nitrogens, the internal nitrogen atoms of purine and oxazole rings (IAR-Ns) and substituted amino groups (OAR-Ns), in polazin Sapanisertib. An interaction between IAR-Ns and under-coordinated Pb2+ at the perovskite crystalline surface is predominant, highly preferable for inhibiting the nonradiative recombination with improving the charge transfer efficiency. The OAR-Ns in Sapanisertib shows little effect on the halide vacancy due to the distribution of charge density. The selective coordination of high-performance passivators with polyazin Sapanisertib can make a boosting power conversion efficiency (PCE) to 18.46%, along with a record certified PCE of 18.27%.
The FP-PSC device is composed of three layers: mesoporous TiO2 (m-TiO2), m-ZrO2 and carbon (Fig. 1a). The experimental details can be found in Supporting information. A drop of isopropanol solution of Sapanisertib was put onto the carbon electrode surface as a modification. The proper concentration of Sapanisertib was optimized according to PCE and photocurrent density-voltage curve (J-V) curve. As shown in Fig. S1 (Supporting information), using the concentration of 0.5 mg/mL Sapanisertib as passivator guaranteed the short-circuit current density (Jsc) and open-circuit voltage (Voc) to reach the maximum, while the filling factor did not change significantly. Fig. 2 showed the field emission scanning electron microscope (FESEM) images of the device surface before and after passivation. The control sample (MAPbI3) and Sapanisertib passivation sample (S-MAPbI3) were well compared. As shown in Fig. 2, compared with the control group (Figs. 2a and c), the S-MAPbI3 (Figs. 2b and d) exhibited denser perovskite layers, fewer grain boundaries and pinholes, which effectively reduces non-radiative recombination due to grain boundary defects. This result is also verified by relevant test results, such as the increase in the open-circuit voltage of S-MAPbI3 from 0.91 V for MAPbI3 to 0.99 V (Fig. 1b). Additionally, the denser crystal layer effectively promotes electron transport, resulting in a partial enhancement of the photocurrent density from 24.33 mA/cm2 in the control group to 24.60 mA/cm2. From the XRD spectrum (Fig. S2 in Supporting information), it can be observed that Sap modification did not significantly change the crystalline shape, indicating that Sap did not enter the crystal lattice of the perovskite. The relatively small increase in the intensity of the 110 plane after Sap modification implies that the Sap modification improved the crystallinity, which is highly consistent with the FESEM results.
Figure 2
Figure 2.
FESEM image of the perovskite solar cell device of the control sample MAPbI3 (a, c) and Sapanisertib passivation sample S-MAPbI3 (b, d).
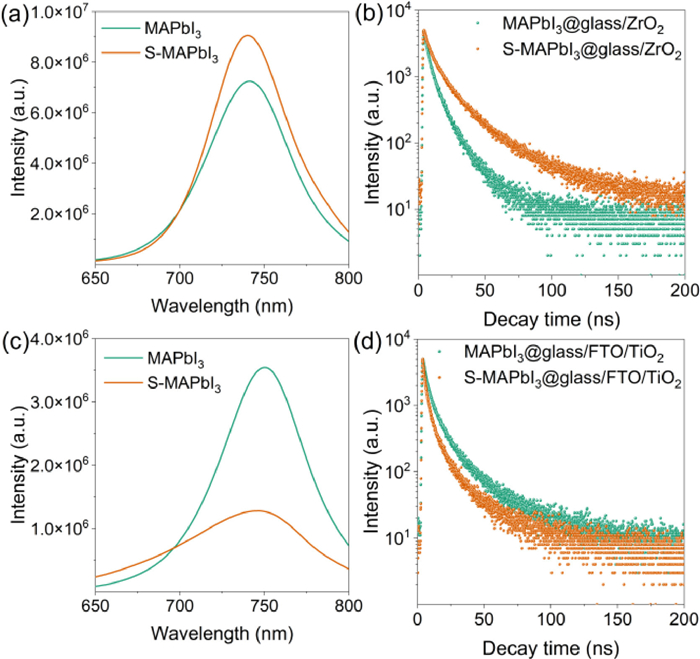
To probe the nonradiative recombination and charge transfer process in MAPbI3 and S-MAPbI3 samples, the photoluminescence and time-resolved photoluminescence spectra were characterized on the insulating substrate (glass/ZrO2, Figs. 3a and b) and conductive substrate (glass/FTO/TiO2, Figs. 3c and d). As shown in Fig. 3a, S-MAPbI3 showed stronger photoluminescence than MAPbI3 on the insulating substrate, meaning that the modification of perovskite film with Sapanisertib molecules inhibits the nonradiative recombination of carriers from the interaction between Sapanisertib and surface defects of perovskite. Moreover, on the conductive substrate (Fig. 3c), S-MAPbI3 sample exhibited lower photoluminescence intensity, indicating that the Sapanisertib modification is beneficial to the charge transmission with enhanced photocurrent density. The time-resolved photoluminescence curves were fitted with Eq. 1:
(1)
Figure 3
Figure 3.
Photoluminescence and time-resolved photoluminescence spectra of the MAPbI3 and S-MAPbI3 modified perovskite films on the insulating substrate glass/ZrO2 (a, b) and conductive substrate glass/FTO/TiO2 (c, d).
where, A is baseline offset constant, B1 and B2 are corresponding attenuation amplitude, τ1 (dominated by surface) and τ2 (mainly led by the internal) are time decay constant [33]. And the calculation results of average attenuation lifetime (τave) are listed in Table S1 (Supporting information) according to Eq. 2:
(2)
On the insulating substrate, from Fig. 3b, the τave is increased from 12.68 ns of MAPbI3 to 16.27 ns of S-MAPbI3, meaning that the repair of defect states by Sapanisertib can produce more excitons under light excitation; On the conductive substrate (Fig. 3d), the τave was decreased from 3.55 ns of MAPbI3 to 1.15 ns of S-MAPbI3, indicative of decreasing internal recombination between electrons and holes in the transport process. These fitting results are consistent with the test results of PL on the different substrates [34].
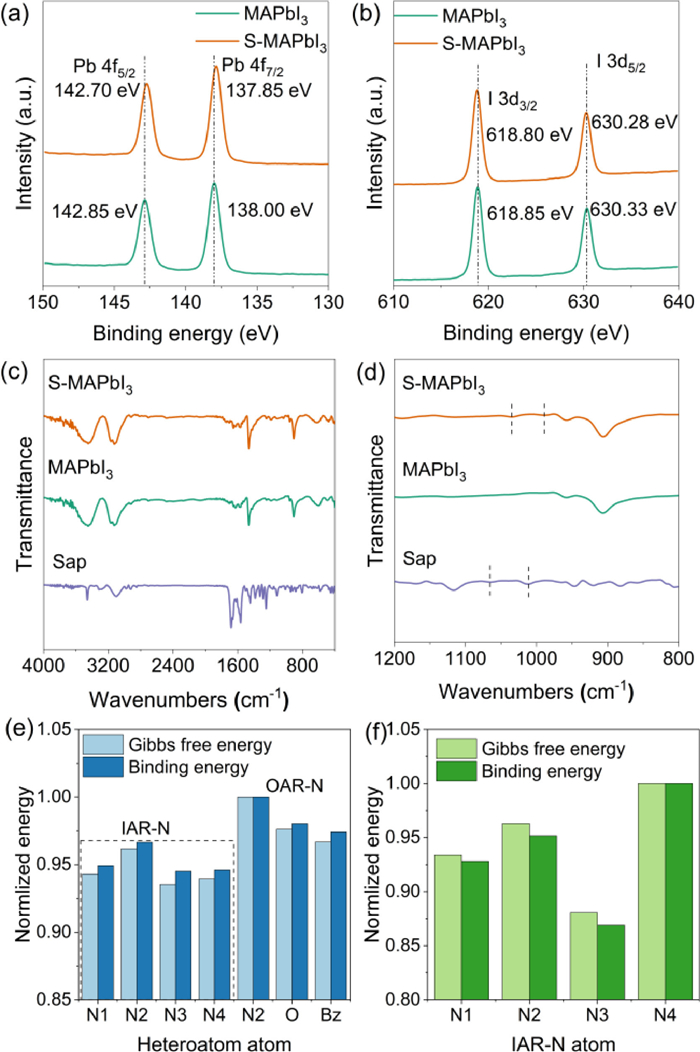
In order to further explore the interaction mode between Sap and perovskite, we first mixed the perovskite precursor and Sap solution with same concentration at 2:1 (v/v) forming perovskite powder sample doped with Sap. Then, X-ray photoelectron spectroscopy (XPS) was performed on this sample. As shown in Fig. 4a, the binding energy of 4f5/2 and 4f7/2 orbitals of Pb in S-MAPbI3 shift from 142.85 and 138.00 eV to 142.70 and 137.85 eV, respectively, compared with MAPbI3. However, the 3d3/2 and 3d5/2 orbits of I (Fig. 4b) only shift from 618.85 and 630.33 eV (MAPbI3) to 618.80 and 630.28 eV (S-MAPbI3), respectively, with an offset value of −0.05 eV. The decrease of binding energy for both of Pb and I indicates increase of electron cloud density around them due to electron donating passivation by Sap. Moreover, in terms of the shift value, the binding effect of Sap with under-coordinated Pb2+ defects in perovskite are stronger than that with iodine inhibiting its migration forming iodine vacancy. As a result, in the passivation process, Sap mainly interacts with the under-coordinated Pb2+ on the surface of the perovskite.
Figure 4
Figure 4.
X-ray photoelectron spectroscopy of MAPbI3 and S-MAPbI3 blends (a) Pb-4f orbit, (b) I-3d orbit. FTIR spectra of MAPbI3, Sap and S-MAPbI3 powders (c, d). The normlized binding energy and Gibbs free energy of some binding sites of Sap to lead atom (e). Normlized Binding energy and Gibbs free energy of four IAR-N atoms in Sap molecule with Pb2+ (f).
In Sap molecule, there are three types of active sites including IAR-N, OAR-N and oxazole-O atom (Fig. 1d). So, which active site interacts with under-coordinated Pb2+? We further tested Fourier transform infrared spectroscopy (FTIR) (Figs. 4c and d). As shown in Fig. 4c, after binding with lead iodide, the vibration of most functional groups in Sap molecule is inhibited. In partially enlarged view (Fig. 4d) of Fig. 4c, the absorption peaks at 1064 and 1015 cm−1 of Sap molecules are red shifted to 1033 and 990 cm−1, respectively, when interaction with perovskite. These peaks come from functional group of Sap molecule because they do not exist for MAPbI3 sample. According to calculated results from density functional theory (DFT) with B3LYP method 6–31 G all electron basis set (Fig. S3 in Supporting information), these peaks are attributed to the rocking vibration of C—H bond and N—H bond on the aromatic ring. To further locate the active site, the complex powders of molecular fragments of Sap, purine and benzoxazole with PbI2 and MAPbI3 were prepared by the same method as Photoluminescence. Their FTIR spectra are presented in Figs. S4-S7 (Supporting information). At the same time, the calculated FTIR spectra of purine and benzoxazole were also carried out by DFT calculations (Figs. S8 and S9 in Supporting information). Comparing Figs. S4-S9, when interacting with PbI2 and MAPbI3, the peak shift in the FTIR spectrum for each sample is owing to the rocking vibration of C—H bond, which is consistent with the results of Sap. Since the N—H bond only exists in OAR-N groups, it indicates that the interaction between Sap and perovskite mainly occurs on the conjugated ring (purine or benzoxazole ring) due to no active site on isopropyl group.
To further clarify the specific action sites on Sap with perovskite, B3LYP method and 6–31G basis set in Gaussian 09 were used to calculate the interaction between Sap and Pb by DFT. First, the surface electrostatic potential of Sap molecule is calculated (Fig. 1c). As shown in Fig. 1c, the positive charge is mainly concentrated around two OAR-Ns, while the negative charge is mainly concentrated around four IAR-N1–4. And the relevant binding energy and Gibbs free energy of C, H, O, N and benzene ring on the Pb atoms is calculated with 6–31 G all electron basis set and lanl2dz pseudopotential basis set and shown in Fig. 4e. The about specific binding energy and Gibbs free energy are listed in Table S3 (Supporting information). It is clear that all IAR-N1–4 have significantly lower binding energy and Gibbs free energy with Pb compared to OAR-N and O. It shows that IAR-N atoms have the greatest binding probability with lead atom, which is the most likely to make up for Pb2+ surface defects.
But the halide defects on the perovskite surface are mainly under-coordinated Pb2+. How do the above atoms behave on Pb2+? As results from same density functional theory calculation, OAR-Ns, benzene ring and oxazole-O do not adsorb at all in the Sap molecular environment, and the corresponding binding energy and Gibbs free energy data cannot be obtained. This is also related to the positive charge distribution around these groups in Fig. 1c. For four nitrogen atoms IAR-N1–4 with highest negative surface potential, the binding energy and Gibbs free energy of N to Pb2+ on the purine ring of IAR-N1–3 are much smaller than those of IAR-N-4 on benzoxazole (Fig. 4f). As a result, the calculation results combined with Pb2+ show that the passivation of under-coordinated Pb2+ can only occur via IAR-N. Furthermore, IAR-N1–3 in purine ring of Sap molecule is more inclined to bind on Pb2+ than IAR-N4 in benzoxazole ring. Combined with the FTIR results, Sap molecule mainly interacts with the exposed lead defect orbital of MAPbI3, and its active site mainly occurs on the IAR-Ns atom in the purine ring, almost does not interact with OAR-Ns. This effect inhibits the nonradiative recombination caused by under-coordinated Pb2+ and improves the carrier transmission efficiency [35,36], thus improving the photoelectric conversion efficiency (Fig. 1b). In addition, since the lone pairs of IAR-N atom participate in the conjugation of the large π bond of purine ring, after interacting with Pb2+, the rocking vibration of C—H bond is affected (Fig. 3d), which further supports the correctness of the calculation results from Figs. 3c and d.
Meanwhile, we conducted 1H NMR tests on the Sap and Sap + PbI2 samples in deuterated DMSO (Fig. S13 in Supporting information). As shown in Fig. S13, the chemical shifts of the d-position hydrogen (H-d) on the purine ring (Fig. 1d) shifted from δ 8.25 and 8.27 to δ 8.29 and 8.30, respectively. The chemical shift moving to a higher field indicates a deshielding effect from the interaction of Pb2+ with IAR-N1–3, lowering the electron density of the purine ring. Regarding the H atoms on the benzene ring, the chemical shift of a-position hydrogen (H-a, Fig. 1d) shifts from δ 7.57 to δ 7.56; and that of H-b shifts from δ 7.27 and 7.25 to δ 7.29, 7.28, 7.27, 7.26, and 7.25. For H-c, from δ 7.49, 7.47, and 7.44 to δ 7.53, 7.52, 7.51, and 7.50. From the shift values, it is clear that the deshielding effect on the benzimidazole ring is smaller than that on the purine ring. A set of peaks at δ 5.11, 5.09, 5.07, 5.06, and 5.04, and another set at δ 1.52 and 1.50 are attributed to the isopropyl group on the purine ring, whose chemical shifts remain relatively constant due to the σ-bonding connecting with the conjugated structure. Therefore, it can be seen that, for the H-d on the purine ring, the interaction of IAR-N1–3 and Pb2+ causes a significant shift to a higher field compared with H-a, b and c due to deshielding effect. This result is in complete agreement with above theoretical calculation results.
To characterize the charge transfer behaviors of the corresponding devices under passivating with IAR-Ns in purine group of Sap, the response curves of voltage (Fig. S12a in Supporting information) and current (Fig. S12b in Supporting information) of FP-PSCs based on MAPbI3 and S-MAPbI3 samples under different light intensities were tested firstly. According to Fig. 5a and Eq. 3, the ideal factors (n) of the devices can be obtained to define their charge recombination type, where n = 1 assumes that recombination occurs only between free electrons and holes in perovskite layer; n approaches 2, defect assisted charge carrier (SRH, Shockley read Hall) recombination is dominant.
(3)
Figure 5
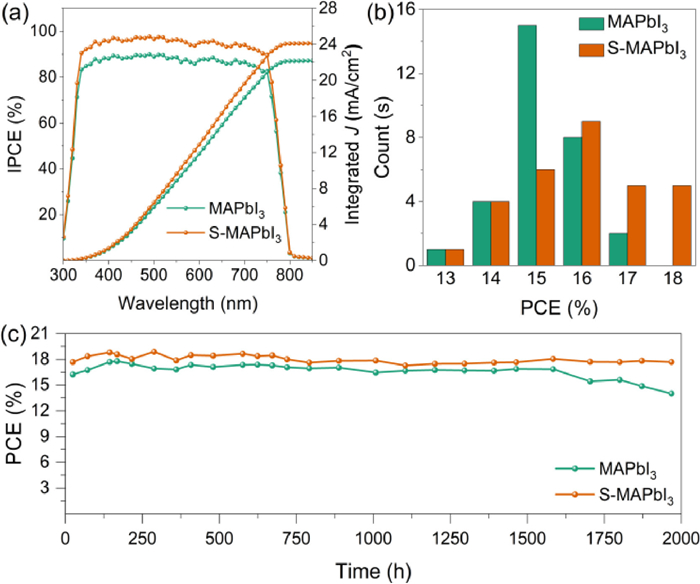
Figure 5.
(a) IPCE curve and its integral current of MAPbI3 devices and S-MAPbI3 devices. (b) PCE distribution histogram of 30 devices. (c) Efficiency stability of the best device in dry air for 2000 h.
where kB is the Boltzmann constant, q is the basic charge, T is the thermodynamic temperature, Plight is the light intensity. Fitting with Eq. 3, the n values of MAPbI3 and S-MAPbI3 samples are 1.18 and 1.07, respectively, indicating the inhibition of SRH recombination by Sap in S-MAPbI3. It is conducive to the improvement of Voc and PCE of the device. For the logarithmic relationship curve between short-circuit current density (Jsc) and light intensity in Fig. 5b, the slopes α of the curves for MAPbI3 and S-MAPbI3 samples can be obtained through double logarithmic coordinate fitting is 0.73 and 0.77, respectively. According to reference, a larger α, a stronger charge extraction capability, which means the increase of charge separation efficiency and charge transfer capability of the device, so that the device shows a higher Jsc [37].
To further determine the modification action of Sap for the defects of perovskite crystals surface, we tested the transient photovoltage (TPV, Fig. S12c in Supporting information) and transient photocurrent (TPC, Fig. S12d in Supporting information) of MAPbI3 and S-MAPbI3 devices. As shown in Fig. S12c, we fit the test results using a single exponential decay equation (f(x) = A + Bexp(-t/τ)) to compare MAPbI3, and the voltage decay time of S-MPbI3 is improved from 10.84 µs to 15.99 µs. The slower voltage decay rate of S-MAPbI3 shows its stronger inhibition of charge recombination, which is conducive to the increase of the open circuit voltage of the device. Moreover, in Fig. S12d (Supporting information), with a decay time of 3.92 µs for S-MAPbI3 and 12.51 µs for the comparison MAPbI3. The faster photocurrent attenuation speed for S-MAPbI3 compared with MAPbI3 evidences the improvement of carrier transport rate. It is beneficial to increase the short-circuit current density of the device [38].
Furthermore, the steady power output curves of MAPbI3 and S-MAPbI3 are shown in Fig. 5c. The voltage bias of MAPbI3 and S-MAPbI3 devices are 0.75 and 0.76 V respectively. S-MAPbI3 shows predominating stability during the test time of 300 s. And in the Nyquist plot (Fig. S12f in Supporting information) of the electrochemical impedance spectroscopy (EIS) at a voltage bias of 0.9 V, the corresponding semicircle can be attributed to the recombination resistance Rrec of the device. The higher Rrec of S-MAPbI3 means its higher suppression capability of nonradiative recombination of charges [39,40], which is highly consistent with the above conclusions.
According to above results, the interaction between IAR-N and under-coordinated Pb2+ is beneficial to the increase of open circuit voltage (Voc) and short circuit current density (Jsc). As shown in Fig. 1b, the PCE of MAPbI3 and S-MAPbI3 are 17.12% (Voc = 0.91 V, Jsc = 24.33 mA/cm2, FF = 77.33%) and 18.46% (Voc = 0.99 V, Jsc = 24.60 mA/cm2, FF = 76.00%), respectively. For certification efficiency of S-MAPbI3, it is 18.27% (Voc = 0.93 V, Jsc = 22.93 mA/cm2, FF = 86.28%). For both devices, Jsc increases from 24.33 mA/cm2 of MAPbI3 to 24.60 mA/cm2 of S-MAPbI3, and Voc increased from 0.91 V to 0.99 V. It is consistent with the conclusion of theoretical calculation and photophysical characterization.
As for the monochromatic incident photoelectrcial conversion efficiency (IPCE) spectra (Fig. 5a), both of the IPCE response platforms fall between 330 nm and 750 nm due to Sap is unincorporated into the perovskite lattice. Only IPCE values of S-MAPbI3 are higher than that of MAPbI3 in this region. Theoretically, the IPCE integrated value by wavelength with standard monochromatic intensity of AM1.5 G sunlight is equal to the test value of Jsc. In Fig. 5a, the integral currents of MAPbI3 and S-MAPbI3 are 23.64 mA/cm2 and 24.12 mA/cm2, respectively. Compared with test Jsc, the lower integrated values may be due to the different active area for IPCE and J-V test, and the charge transfer rate of the device is not proportional to the light intensity.
Meanwhile, S-MAPbI3 also shows better efficiency distribution (Fig. 5b). 30 devices tested for MAPbI3 and S-MAPbI3 show similar normal distribution. The PCE values of the former are more distributed between 15%~16%, while that of latter are mostly higher than 16%. The relevant statistics of Voc, Jsc, FF and PCE are presented in Fig. S10 (Supporting information). S-MAPbI3 still demonstrates its advantages. For the long-term operation stability in dry air of the devices, the PCE of S-MAPbI3 still maintain more than 90% of its initial value after 2000 h, while that of MAPbI3 control group has decreased to less than 80% of its initial value (Fig. 5c).
In this study, we utilized different chemical environments of nitrogen atoms in the anticancer drug Sapanisertib molecule, specifically IAR-N located on the purine and benzoxazole rings and OAR-N located on the -NH2, to achieve selective passivation of the perovskite surface. Our hypothesis, supported by X-ray photoelectron spectroscopy and Fourier transform infrared spectroscopy, is that the selective adsorption of under-coordinated Pb2+ in perovskite occurs through IAR-N located on the benzoxazole and purine rings. Density functional theory calculations revealed that IAR-N located in the ring has a lower binding energy and Gibbs free energy for Pb2+. Our findings were further supported by 1H NMR spectroscopy, which showed that the H in the purine ring was significantly more electronically shielded than the H in the benzoxazole ring. Moreover, the introduction of Sap molecules improved both crystallization formation and electron transport in perovskite, leading to a record certified PCE of 18.27% (and up to 18.46%). This work provides a useful guide for screening organic small molecule passivators, which is crucial for reducing inherent defects and improving the photovoltaic conversion efficiency of printable mesoscopic perovskite solar cells.
Declaration of competing interest
The authors declare that they have no financial and personal relationships with other people or organizations that can inappropriately influence the work.
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (Nos. 21421004 and 22075083), the Programme of Introducing Talents of Discipline to Universities (No. B16017).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108629.
[1]
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 132 (2010) 154104. doi: 10.1063/1.3382344
[2]
R.E. Brandt, V. Stevanović, D.S. Ginley, T. Buonassisi, MRS Commun. 5 (2015) 265–275. doi: 10.1557/mrc.2015.26
Figure 1
Structural information of the Sapanisertib molecule and its action process in FP-PSCs. (a) Schematic illustrations of IAR-N passivation for under-coordinated Pb2+ in perovskites over OAR-N in the Sapanisertib molecular environment. (b) J-V curve of MAPbI3 devices and S-MAPbI3 devices. (c) Molecular surface electrostatic potential of Sapanisertib. (d) Chemical structure and different kinds of heteroatoms of Sapanisertib.
Figure 3
Photoluminescence and time-resolved photoluminescence spectra of the MAPbI3 and S-MAPbI3 modified perovskite films on the insulating substrate glass/ZrO2 (a, b) and conductive substrate glass/FTO/TiO2 (c, d).
Figure 4
X-ray photoelectron spectroscopy of MAPbI3 and S-MAPbI3 blends (a) Pb-4f orbit, (b) I-3d orbit. FTIR spectra of MAPbI3, Sap and S-MAPbI3 powders (c, d). The normlized binding energy and Gibbs free energy of some binding sites of Sap to lead atom (e). Normlized Binding energy and Gibbs free energy of four IAR-N atoms in Sap molecule with Pb2+ (f).
Figure 5
(a) IPCE curve and its integral current of MAPbI3 devices and S-MAPbI3 devices. (b) PCE distribution histogram of 30 devices. (c) Efficiency stability of the best device in dry air for 2000 h.

 DownLoad:
DownLoad:


 下载:
下载:






