Citation:
Shuai Cao, Leqian Song, Huacheng Zhang, Jie Han, Yanli Zhao. Self-assembly of partially oxidized pillar[5]arenes induced by intermolecular charge transfer interactions[J]. Chinese Chemical Letters,
2023, 34(12): 108479.
doi:
10.1016/j.cclet.2023.108479
Self-assembly of partially oxidized pillar[5]arenes induced by intermolecular charge transfer interactions
English
Self-assembly of partially oxidized pillar[5]arenes induced by intermolecular charge transfer interactions
zhaoyanli@ntu.edu.sg (Y. Zhao). 1 These authors contributed equally to this work.
Received Date:
09 February 2023 Accepted Date:
17 April 2023 Revised Date:
05 April 2023 Available Online:
15 December 2023
Abstract:
Supramolecular interactions such as π-π stacking interaction and charge transfer interaction have drawn much attention in the design and construction of various supramolecular assemblies. Herein, partially oxidized pillar[5]arene (P5A), pillar[4]arene[1]quinone (P4A1Q), pillar[3]arene[2]quinone (P3A2Q), and pillar[2]arene[3]quinone (P2A3Q) were synthesized by one-step reaction. As indicated by experimental characterization data and density function theory modeling results, charge transfer interaction among partially oxidized P5A plays a significant role in host-host self-assembly behavior and corresponding packing morphology. This work provides a unique strategy for the construction of functional macrocyclic assemblies through host-host self-assembly.
Supramolecular chemistry witnesses impressive advances [1-4] in aspects such as molecular recognition [5,6] and self-assembly [7-9], affording the primary objective to create assemblies composed of various building blocks from inorganic elements [10,11] to organic molecules [12-16] through the noncovalent bonding including π-π stacking [17,18], hydrogen bonding [17-20], hydrophilic/hydrophobic [7,21-23], host-guest [5,24-27] and charge transfer (CT) interactions [13,17,19,24-32]. Among these weak interactions, CT interaction has received significant attention in recent years due to the valuable contribution toward oxidative phosphorylation [33,34] in organism and prospective applications in organic electronics [27], organic photovoltaic devices [35,36] and storage materials [2]. CT interaction consisting of intramolecular and intermolecular forms depends on the couple of highest occupied molecular orbital (HOMO) of π-donor and the lowest unoccupied molecular orbital (LUMO) of π-acceptor in two distinct and somewhat separable regions [37,38]. However, single-molecule organic systems with intramolecular through-space CT often require complicated syntheses and multiple fabrication processes [5,27,37,38]. On the other hand, most of intermolecular CT self-assemblies (CTSAs) in supramolecular field usually co-exist with host-guest interactions on account of the formation of donor and acceptor pairs [5,37], while host-host intermolecular CT interaction is often neglected under the cover of host-guest chemistry. Therefore, the study of CTSAs based on macrocyclic host molecules is challenging but worth deep investigations.
As a representative macrocycle, pillar[n]arene is well-known for the unique symmetrical pillar-like architecture, being as one of the latest generation supramolecular hosts [39,40]. Pillararene-based CTSAs based on host-guest interaction have been developed especially via “exo-wall” interactions [29,30] and intermolecular hydrogen bonding interaction [19,20,41], while host-host CTSAs involving pillararenes have been less studied. Several practical studies on pillararene-based CTSAs were achieved through inter/intramolecular hydrogen bonding interaction. For example, by adjusting the charging ratio, Ogoshi et al. reported that the co-assembly of pillar[5]quinone (P5Q) and hexagonal pillar[6]arene (P6A) favors constructing spherical architectures [17]. Kaifer et al. discovered that partially oxidized P5A possessing p-benzodiquinone and p-dihydroxybenzene could self-assemble into fibrous structures driven by intramolecular hydrogen bonding interaction [19]. Recently, Yang et al. reported the host-host CT crystallization utilizing the eutectic contacting reaction in 4 ℃, where p-benzodiquinone of P5Q served as the electron acceptor and 1,4-dimethoxybenzene of P5A acted as the electron donor [29]. In comparison with P5A, it is anticipated that host-host CT interaction might appear in partially oxidized P5A skeleton consisting of p-benzodiquinone and 1,4-dimethoxybenzene subunits. The structure of corresponding self-assemblies can be varied, depending on the strength variations between π-π stacking and CT interactions. Meanwhile, the study of host-host CT interaction in partially oxidized P5A could provide a feasible inspiration for constructing diverse host-host CTSAs in supramolecular macrocyclic chemistry.
In this work, we propose the conception of host-host CT interaction in partially oxidized P5A, and report the syntheses of several partially oxidized pillararene derivatives, i.e., pillar[5]arene (P5A), pillar[4]arene[1]quinone (P4A1Q), pillar[3]arene[2]quinone (P3A2Q), and pillar[2]arene[3]quinone (P2A3Q), via one-step strategy. Both theoretical simulations and experiment results prove that the CT interaction is a major driving force in the self-assembly process of these host structures (P4A1Q, P3A2Q and P2A3Q). Compared to P5A, with the increase in the quantity of p-benzodiquinone, the CT interaction in P4A1Q, P3A2Q and P2A3Q is dramatically enhanced, leading to the change of their morphologies from cubic arrangement to fragments and finally long fibrous shape. Meanwhile, density function theory (DFT) studies also elaborate that the stacking modes of these host dimers exhibit a profound influence on intra/inter-host CT interactions.
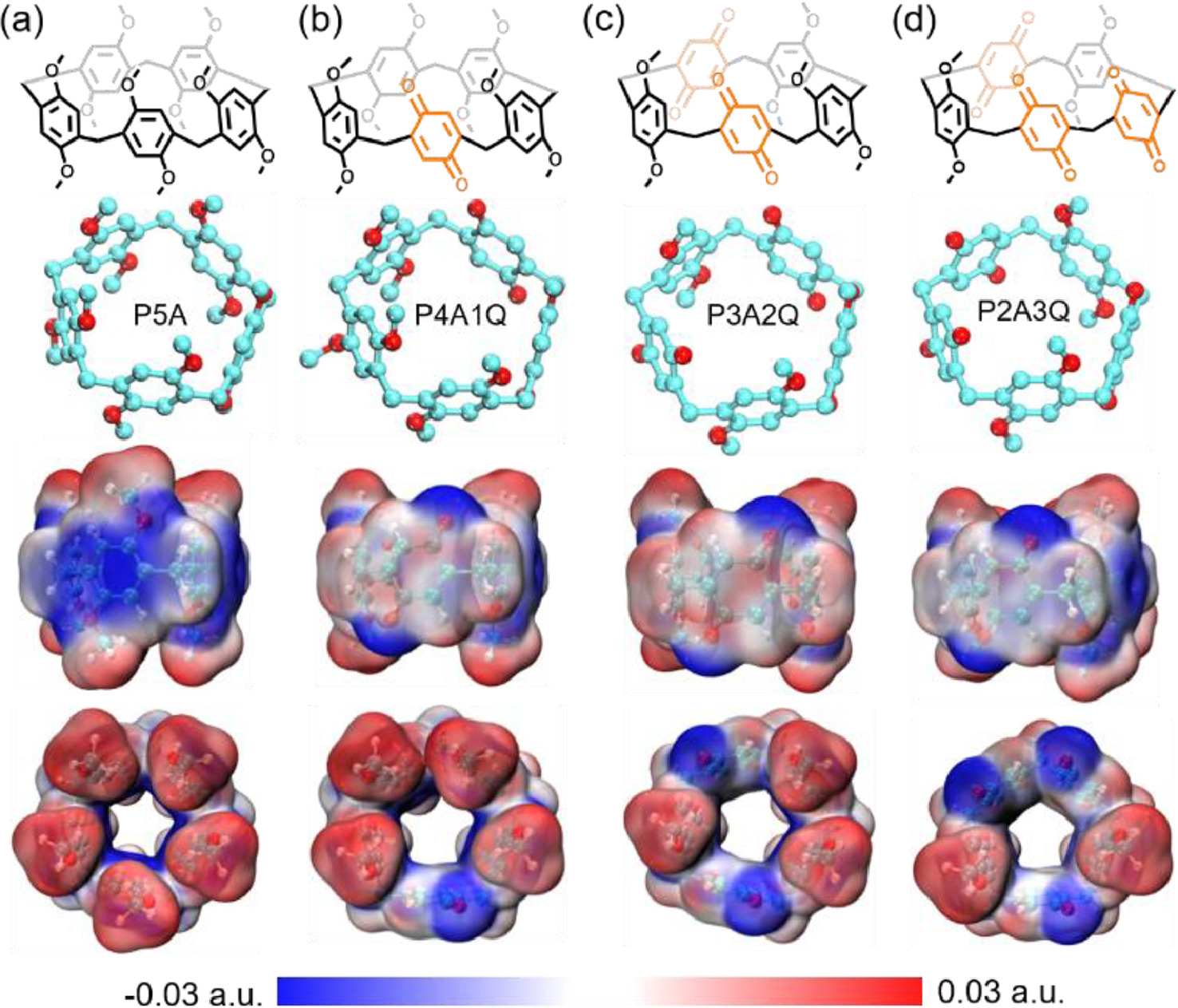
The macrocyclic structure of P5A is named for its electron-rich cavity with a conjugated pillar-type structure (Fig. 1a), leading to the production of π-π stacking interactions between neighboring P5A hosts [18]. In partially oxidized P5A, such as P4A1Q, P3A2Q and P2A3Q, the presence of electron-withdrawing moiety, i.e. p-benzoquinone, was of great value to the clear change in space charge distribution (Figs. 1b-d), offering an unique opportunity to investigate the “exo-wall” interactions that are different from conventional π-π stacking interactions in P5A.
Figure 1
Figure 1.
Chemical structures and electrostatic potential maps (side view and top view) of four macrocyclic molecules. (a) P5A, (b) P4A1Q, (c) P3A2Q and (d) P2A3Q. Note: The structures at the second row involve the computational model of macrocyclic molecules used in the DFT studies. Blue region represents low potential with the abundance of electrons. Red region represents high potential with the relative absence of electrons.
Comparing P5A to P4A1Q, P3A2Q and P2A3Q, there is mainly π-π stacking interactions between P5A hosts, and the primary self-assembly pattern between them might be different and influenced by possible CT interaction. Given the self-assembly process, inter/intramolecular CT interactions should be expected. In fact, intramolecular CT interaction existed in partially oxidized P5A has been suggested in some pioneering research studies [19,41-43]. Then, we were very curious about two following questions: Is there intermolecular CT interaction in partially oxidized P5A in comparison with P5A, and what are the visible effects of intra/intermolecular CT interactions on the crystalline morphology and the behavior of host-host-based self-assembly?
Initially, P5A and partially oxidized P5A including P4A1Q, P3A2Q and P2A3Q were prepared by adjusting the conventional synthesis methods (Scheme S1 in Supporting information) [44-47]. It was found that tetrahydrofuran is unnecessary during the synthesis procedure, and three oxidation products can be obtained simultaneously in one-step reaction. The yields of compounds P4A1Q, P3A2Q and P2A3Q were 42.22%, 14.20% and 3.48%, respectively. The structures of target products were fully characterized and confirmed by 1H NMR, high-resolution mass spectrometry (HR-MS) and 13C NMR (Figs. S1-S10 in Supporting information) [44,48,49]. Although P3A2Q and P2A3Q have different constitutional isomers, only one type of constitutional isomer was obtained by our methods. It may be the request of the lowest energy principle that p-benzoquinone units sit as far from each other as possible to weaken the Coulomb repulsion interactions. On the other hand, the influence of different solvents on the products may also be considered.
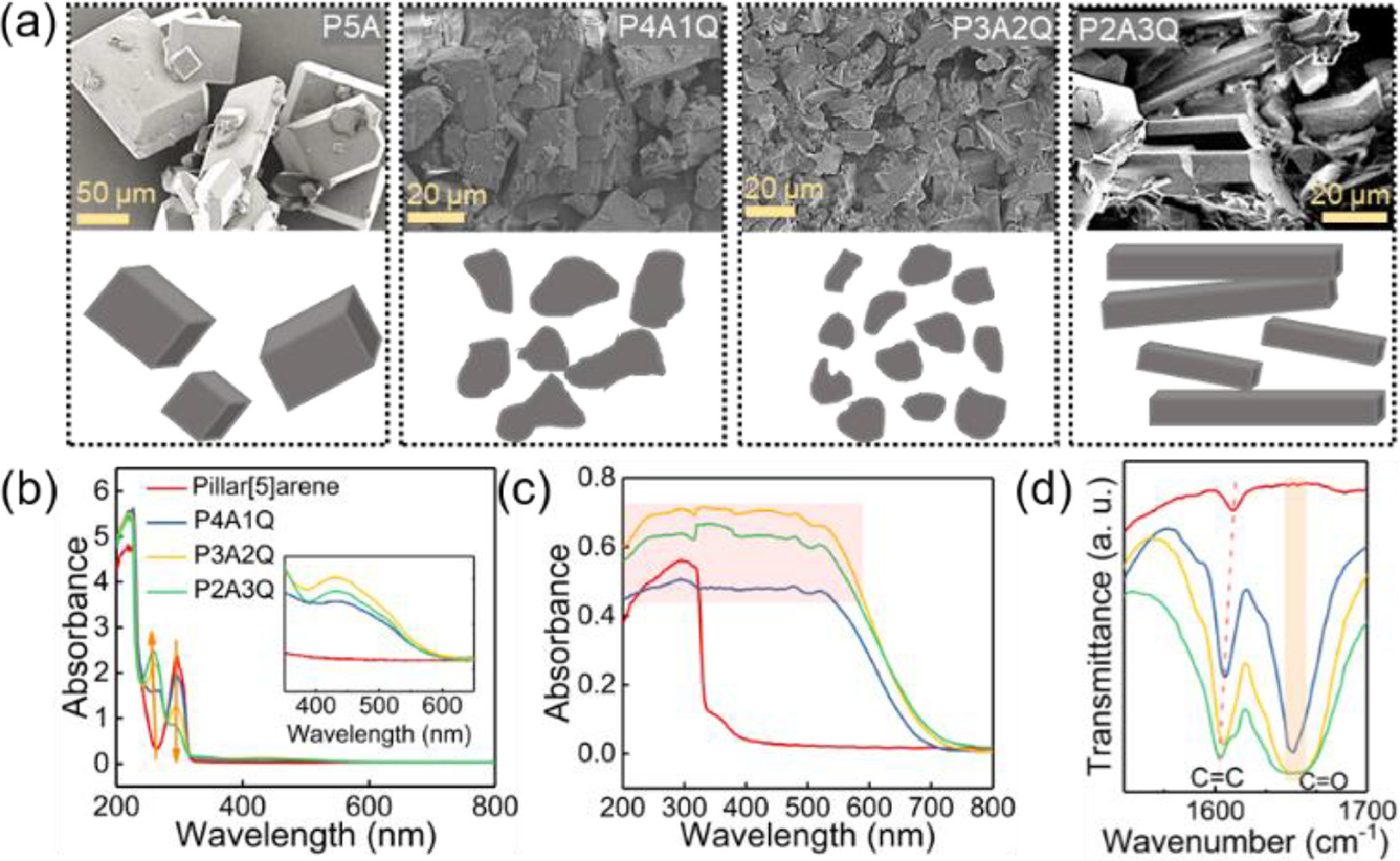
Subsequently, the morphology difference between P5A and partially oxidized P5A driven by CT interaction was investigated by scanning electron microscopy (SEM, Fig. 2a). P5A gives typically cubic crystals, while the morphology of P4A1Q, P3A2Q and P2A3Q undergoes a remarkable change from fragment to the final long fibrous shape. SEM images also indicate that the sizes of crystalline P4A1Q and P3A2Q are around 20 µm, and the length of needle-like P2A3Q is greater than 20 µm with the width of around 5 µm. Compared to partially oxidized P5A, the crystalline grains of P5A are more homogenous and regular, thus meaning that CT and π-π stacking interactions are competitive during the self-assembly process.
Figure 2
Figure 2.
Compositional and morphological characterization of four macrocyclic molecules. (a) SEM images and schematic illustration of morphological evolution; (b) UV-vis spectra (0.01 mmol/L in dichloromethane); (c) UV-vis DRS spectra; (d) Partial FT-IR spectra of P5A P4A1Q, P3A2Q and P2A3Q.
Optical spectroscopic measurements were performed to examine the feasibility of CT interaction in partially oxidized P5A. The UV-vis spectra (Fig. 2b) of P5A, P4A1Q, P3A2Q and P2A3Q (0.1 mmol/L) were detected in dichloromethane solution. With the increase of oxidation degree, the absorption peak corresponding to benzoquinone unit located at 240–260 nm region becomes stronger, while absorption intensity associated to 1,4-dimethoxybenzene unit in the 290–300 nm region decreases [19,42]. Obvious CT absorption peak in the 400–650 nm region could also be observed in enlarged figure (Fig. 2b). It is noteworthy that the intensity of CT band in the 400–650 nm region for partially oxidized P5A rises (Fig. S11 in Supporting information) [41-43], whereas P5A shows no absorbance in this region, clearly revealing the formation of CT interaction in partially oxidized ones.
Additionally, a broad CT band of P4A1Q, P3A2Q and P2A3Q spreading over the entire spectra (Fig. 2c) can be observed in UV-vis diffuse reflectance spectra (DRS) when compared with that of P5A, indicating intermolecular CT interaction between the donor hydroquinone unit and the acceptor benzoquinone unit [19,29,30]. FT-IR spectra (Fig. S12a in Supporting information) confirmed that methylene and methoxy units remained, as shown in the absorptions at 2939 cm−1 and 1213 cm−1 [50,51]. As for partially oxidized P5A, an apparent absorption peak at 1549 cm−1 related to C═O can be found (Fig. 2d) [52]. Meanwhile, the absorption band assigned to C═C of the benzene skeleton stretching red-shifts gradually (Fig. 2d), suggesting the CT interaction between hydroquinone and benzoquinone units as well [53].
As depicted in Fig. S12b (Supporting information), powder X-ray diffraction (XRD) analysis shows multiple crystalline phases in P5A and partially oxidized P5A. The crystal phase patterns of P4A1Q, P3A2Q and P2A3Q are similar, but P5A has more uniformed size and better crystallinity without the interference of CT interaction. Then, their pyrolysis character and thermostability were analyzed comprehensively based on thermogravimetric analysis (TGA, Fig. S12c in Supporting information). It was noticeable that P5A has better thermal stability than partially oxidized P5A, due to active carbonyl group. As shown in Fig. S13 (Supporting information), the bandgap energy values for P5A, P4A1Q, P3A2Q and P2A3Q are 3.77 eV, 1.76 eV, 2.06 eV and 2.08 eV, respectively. In comparison with P5A, partially oxidized P5A hosts display smaller bandgap energy and thus possess a higher electron transmitting capacity resulting from intra/intermolecular CT interactions [19].
With the concept of nonporous adaptive crystals (NACs) being put forward, pillararene-based self-assembled materials play a major role in selective adsorption of organic molecules in the solid state [29,30]. Absorption characteristics of P5A, P4A1Q, P3A2Q and P2A3Q were thoroughly investigated by nitrogen adsorption-desorption isotherms (Fig. S14 in Supporting information). The Brunauer–Emmett–Teller (BET) surface area of the samples was very small and all about 2 m2/g, which may be the primary reason that the hysteresis loop is asymmetry [54]. Although they possess small specific surface area, they could be employed as NACs to provide specific adsorption of different organic molecules.
To further demonstrate the host-host CT interactions in partially oxidized pillararene derivatives, a series of control experiments using p-benzodiquinone (p-BQ) monomer and 1,4-dimethoxybenzene (p-DMB) was also carried out (Fig. S15 in Supporting information). With the increase of p-DMB and p-BQ content, the intensity of CT peak of partially oxidized pillararene became stronger, while that of P5A remained roughly the same with the increase of p-DMB content. These results reveal that P4A1Q, P3A2Q and P2A3Q possess the properties of both charge donor and acceptor, thus building the foundation of intermolecular CT interactions. Furthermore, the solvent dependence of the intermolecular CT process was also validated in dimethylformamide, methanol, acetonitrile, acetone, ethyl acetate and 1,2-dichloroethane, from higher polar solvents to lower ones (Fig. S16 in Supporting information). It is noteworthy that a weaker CT effect could be observed in methanol due to the low solubility of partially oxidized pillararene.
For a better insight into different stacking modes of P4A1Q, P3A2Q and P2A3Q units induced by CT and π-π stacking interactions, 1H–1H nuclear overhauser effect spectroscopy (NOESY) NMR (Figs. S17 and S18 in Supporting information) and correlation spectroscopy (COSY) NMR (Fig. S19 in Supporting information) were performed, which was employed to efficiently illustrate the corresponding interaction between neighboring protons in different types of benzene rings and simplify the analysis of the stacking arrangements of their dimer assemblies [30]. Due to similar information given by NOESY NMR and COSY NMR spectra, intramolecular proton-proton interactions and CT interactions are of course correct in partially oxidized P5A. A combination of the above experiment confirmation of the existent host-host CT interactions in partially oxidized pillararene would provide a glimpse that how macrocycle dimers self-assemble as a new mode is acceptable. The corresponding peak of H1-H1 indicates that weak π-π stacking interaction between nearby benzene rings originates from the self-assembly of two neighboring P5A, as shown in Fig. S17a (Supporting information). Other corresponding peaks such as H1-H2 and H1-H3 appeared in Fig. S17b (Supporting information) reveal different stacking modes of P4A1Q from P5A, due to the possession of CT interaction. Similar phenomena could be observed in the case of P3A2Q and P2A3Q, with corresponding peaks such as H1-H3 in Figs. S17c and d (Supporting information). The main assembly modes were analyzed in detail (Fig. S20 in Supporting information) by altering the relative positions of p-benzodiquinone and 1,4-dimethoxybenzene subunits.
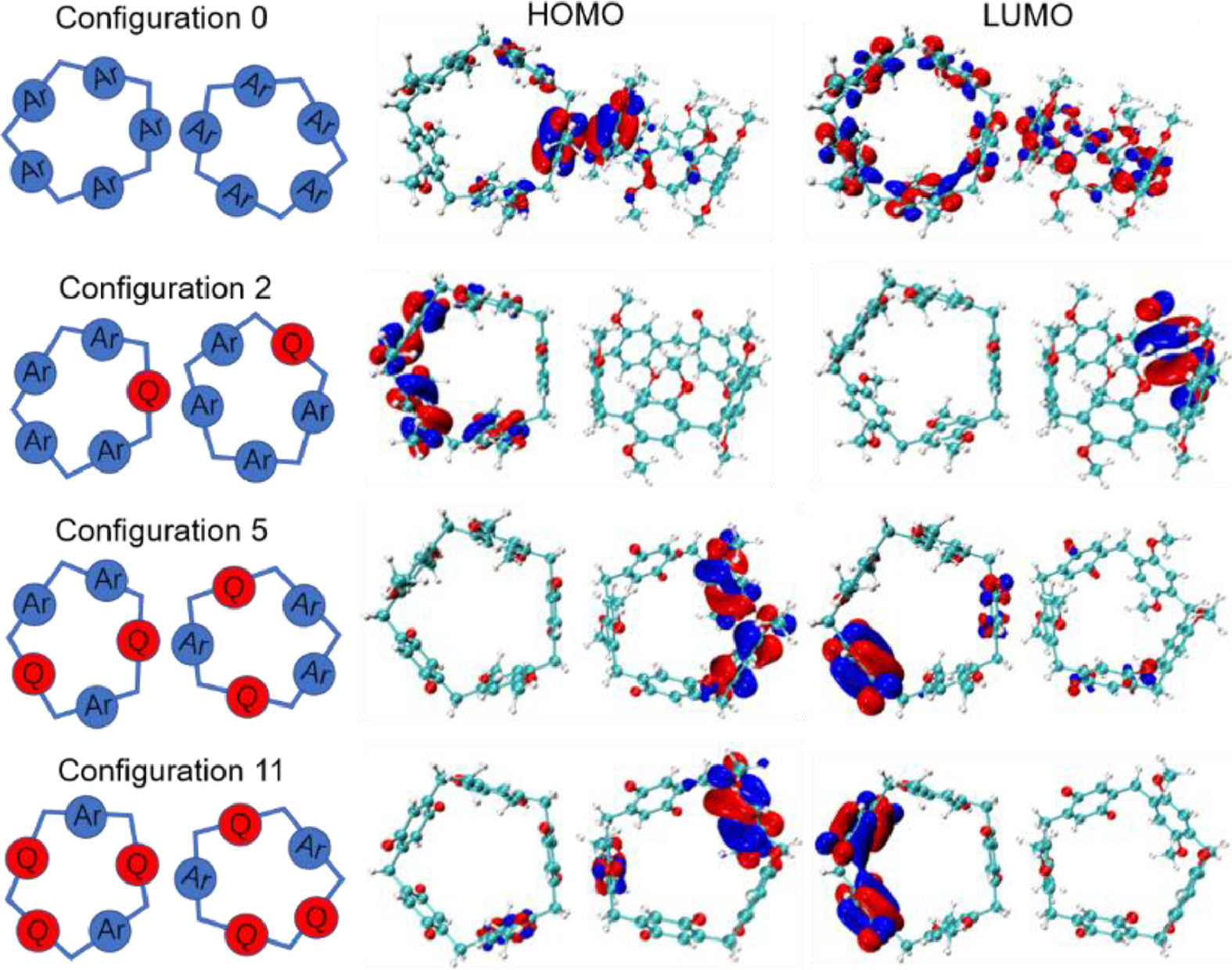
Subsequently, density function theory (DFT) computational studies of macrocycle dimers were carried out to evaluate these configurations from the view of quantum chemistry [19,42]. The M062X/6–31G(d,p) energies for the optimized dimer self-assemblies of four compounds were shown in Table S1 (Supporting information). The calculated Gibbs free energy of the fourteen configurations containing the same dimer has the same value, while HOMO-LUMO energy gap and the distribution of HOMO/LUMO (Figs. S21 and S22 in Supporting information) obtained via Multiwfn code are different. As for P5A dimer-based self-assemblies, electron-rich area resulted from π-π stacking interaction contributed much to HOMO and LUMO has a large diffused density distribution over the molecular skeleton, in which exo-wall CT interaction is impractical [19]. Because LUMO and HOMO are located on separate monomers and consequently intermolecular CT interaction is expected, some typical configurations in favor of the occurrence of intermolecular CT interaction could be further exhibited in Fig. 3. In other stacking modes including configurations 4, 8, 10 and 13, LUMO and HOMO are distributed on one of the monomers, promoting intramolecular CT interaction. As an indicator of the stability of the reaction products, ΔELUMO-HOMO values indicate that the configurations 3, 5, 6, 10, 11 and 12 are the stable formations of dimers, promoting the further analysis of these configurations [55].
Figure 3
Figure 3.
Frontier molecular orbitals for dimers of P5A, P4A1Q, P3A2Q and P2A3Q in accordance with configurations 0, 2, 5, and 11.
The interaction energy of each assembly was obtained from wavefunction analyses of independent gradient model based on Hirshfeld partition (IGMH) [56]. In general, hydrogen bonding interaction between oxygen atom on C═O group and hydrogen atom on -CH3 plays a vital role for the stabilization of various configurations (Fig. S23 in Supporting information). For the case of P3A2Q dimers, configuration 4 is unstable, because a long-range repulsive force can be seen when two p-benzoquinone subunits getting closer to each other. Configurations 5 and 6 are more stable, benefiting from intermolecular CT interaction. With the same reference in mind, configuration 3 is optimal, because intermolecular and intramolecular CT interactions would occur simultaneously. Configurations 10, 11 and 12 of P2A3Q dimers are more stable, as the main intermolecular CT interaction is associated with these configurations. Thus, packing modes indicated from computational calculations are consistent with the analysis results of 1H–1H NOESY NMR spectra.
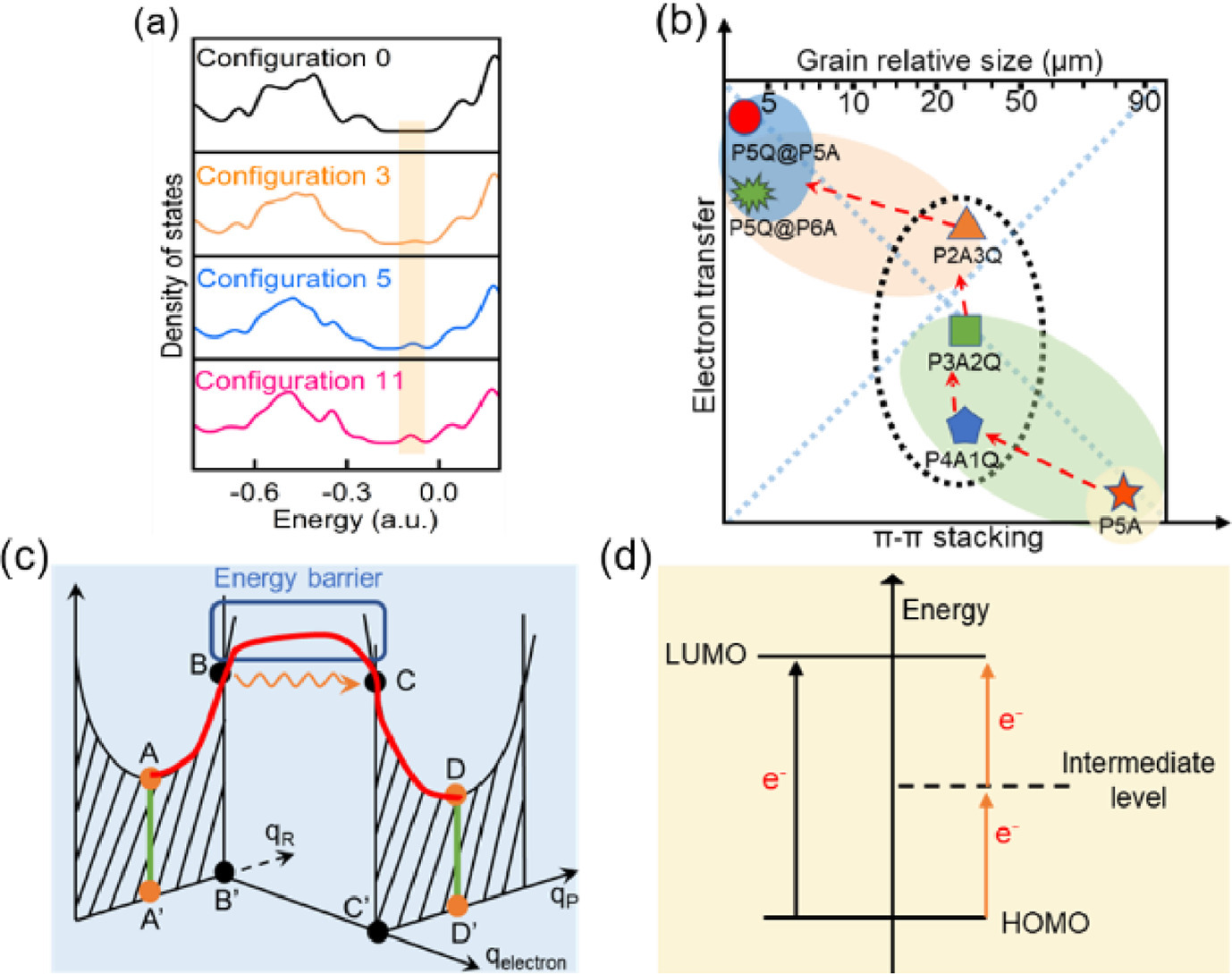
Density of states (DOS) were obtained via Multiwfn code combining with the visual molecular dynamic (VMD) program to study the CT of various dimer-based self-assemblies [57]. As shown in Fig. 4a and Fig. S24 (Supporting information), new states appear near the Fermi level of P4A1Q, P3A2Q and P2A3Q dimer units, which might be responsible for the enhanced intermolecular CT interaction. For the purpose of comparison, P1A4Q was tested by the analogy calculation (Figs. S25-S27 in Supporting information). Among five configurations, configuration 1 and configuration 5 are most stable (Table S2 in Supporting information). Significantly, Coulomb repulsion between two p-BQ could be effectively reduced by the rotation of benzene rings in configuration 5 and two new states appear near the Fermi level at the same time. All these factors could facilitate the intermolecular CT interaction.
Figure 4
Figure 4.
Schematic diagram of charge transfer. (a) calculated DOS of configurations 0, 3, 5 and 11. The isosurface is 0.004 a.u. (b) Morphological changes of different pillararene-based systems, from P5A (strong π-π stacking), P4A1Q/P3A2Q/P2A3Q to P5Q@P6A and P5Q@P5A (strong CT). (c) Charge transfer based on Frank-Condon principle. (d) Charge transfer between LUMO and HOMO.
As for pillararene-based systems with strong CT such as P5Q@P6A [17] and P5Q@P5A [29], their morphology usually displays regularly ordered crystals with smaller sizes (Fig. 4b). The foregoing analysis shows that the stacking modes of these host dimers vary, mainly because of the balance between CT and π-π stacking interactions (Fig. S28 in Supporting information). The competition of these two interactions are the major factors on specific disorder and fragmental morphology.
According to Franck-Condon principle [58,59], a higher orbital overlap between electron acceptor A and electron donor D means a lower electron tunneling energy barrier, and thus the greater the probability of CT (Fig. 4c). It is understandably easy that the distribution of LUMO and HOMO as well as the interaction energy could influence intermolecular CT interaction because they could impact the probability and tunneling energy barrier of CT, respectively. Thus, configurations 3, 5 and 11 could effectively promote intermolecular CT. Meanwhile, new states between LUMO and HOMO could increase orbit coupling as well, facilitating the CT (Fig. 4d).
In conclusion, we have demonstrated the generated self-assembly forms and stability mechanism of host-host CT interaction by using partially oxidized P5A (P4A1Q, P3A2Q and P2A3Q) based on experiment studies and theoretical calculation. The differences in morphology and pore distribution of these CTSAs, as well as competition between host-host CT and π-π stacking interactions have been investigated in detail, revealing that the stable conformations of P3A2Q and P2A3Q dimers exist in the form of intermolecular CT from HOMO composed of 1,4-dimethoxybenzene subunits to LUMO containing p-benzoquinone moieties. Meanwhile, the present of new electronic states is beneficial for the transfer of intermolecular charges. This work not only offers an efficient strategy to build macrocycle-based CT complexes, but also provides an interesting perspective for reasonable regulation and control of hierarchical supramolecular self-assemblies.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
H. Zhang acknowledges the financial support from the “Young Talent Support Plan” (No. 050700-71240000000046) of Xi’an Jiaotong University and Natural Science Foundation of Shaanxi Province (No. 2021JM-006). J. Han acknowledges the 111 Project (No. B12015).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108479.
N.S. Bayliss, E.G. McRae, J. Phys. Chem. A 58 (1954) 1002–1006. doi: 10.1021/j150521a017
Figure 1
Chemical structures and electrostatic potential maps (side view and top view) of four macrocyclic molecules. (a) P5A, (b) P4A1Q, (c) P3A2Q and (d) P2A3Q. Note: The structures at the second row involve the computational model of macrocyclic molecules used in the DFT studies. Blue region represents low potential with the abundance of electrons. Red region represents high potential with the relative absence of electrons.
Figure 2
Compositional and morphological characterization of four macrocyclic molecules. (a) SEM images and schematic illustration of morphological evolution; (b) UV-vis spectra (0.01 mmol/L in dichloromethane); (c) UV-vis DRS spectra; (d) Partial FT-IR spectra of P5A P4A1Q, P3A2Q and P2A3Q.
Figure 4
Schematic diagram of charge transfer. (a) calculated DOS of configurations 0, 3, 5 and 11. The isosurface is 0.004 a.u. (b) Morphological changes of different pillararene-based systems, from P5A (strong π-π stacking), P4A1Q/P3A2Q/P2A3Q to P5Q@P6A and P5Q@P5A (strong CT). (c) Charge transfer based on Frank-Condon principle. (d) Charge transfer between LUMO and HOMO.

 DownLoad:
DownLoad:

 下载:
下载:





