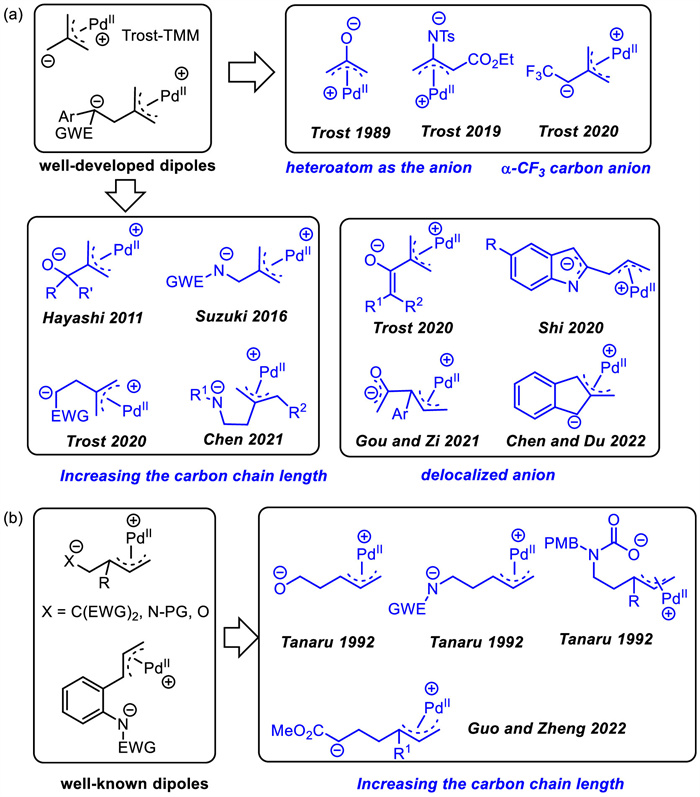
Scheme 1.
Representative Pd-π-allyl zwitterions.

Dipolar cycloaddition reactions are of fundamental importance to organic synthesis,as they provide access to cyclic motifs in an efficient and convergent manner,particularly,the capability to rapidly access fused-/bridged-ring systems and medium-sized rings (7 to 11-membered rings) which are common core structures in various bioactive natural products and drugs [1-11]. As a result,this strategy has attracted the attention of numerous synthetic chemists. In addition to relatively stable classical 1,3-dipoles [12-15],1,n-dipole variants which are not fully conjugated chemical species,are difficult to generate using non-catalytic methods. Among them,transition metal Pd-catalyzed dipolar cycloadditions have been one of the most straightforward and powerful strategy to this end. Most of dipolar intermediates are a short-lived,nonisolable species and the dipole species could be a zwitterion rather than a true dipole. In 1980s,the best-known palladium-mediated dipole,palladium-trimethylenemethane (Pd-TMM),was firstly generated by Trost's group [16],which is an all-carbon 1,3-dipole. Thereafter,Shintani and Hayashi et al. designed γ-methylene-δ-pentyl lactone as a precursor of all-carbon 1,4-dipole compound to access larger ring sizes (Scheme 1a) [17,18]. It has been used extensively in the preparation of a variety of cyclic products,since it reacts with a multitude of acceptors. With the rapid development of these early studies,increasingly profound dipoles have been emerged and are widely used (Scheme 1a). As shown in Scheme 1a,compared to Pd-TMM dipoles,those new dipoles are designed by introducing heteroatoms,growing carbon chains or designing dipoles containing delocalized anions,which not only allow to construct a larger ring system,but also have more reaction pathways for cyclization reactions. This type of dipolar reaction is characterized by the formation of exocyclic unsaturated bonds.
Around the same time as the development of the [3 + 2] Pd-TMM cycloadditions,an alternative [3 + 2] cycloaddition strategy was realized by Tsuji and co-workers [19] via a zwitterion generated in situ by the reaction of Pd with activated vinylcyclopropanes. Since then,the Pd-catalyzed intermolecular [3 + n] and [5 + n] cycloadditions have been a powerful,atom-economic tool for the construction of various carbon- and heterocycles through the in situ generated Pd-π-allyl 1,3- or 1,5-zwitterions by the reaction of Pd(0) with vinyl three-membered cycles and their analogues [20-30]. Meanwhile,Pd-π-allyl zwitterions derived from vinyl benzoxazinanones were first developed as a type of 1,4-dipole by Tunge's group in 2008 [31],and subsequently gained widespread use [32-39]. Several related reviews have been reported (Scheme 1b) [18,40].
In recent years,along with the expansion and applications of those dipoles,some other dipoles that had little application after their initial discovery have recently received renewed attention in palladium-catalyzed cycloaddition reactions and have been rapidly developed (Scheme 1b). Compared to dipoles derived from vinyl three-membered cycles,these newly applied dipoles have longer chains to enable larger cyclization products. It is worth noting that the reaction products usually containing multiple chiral centers can be constructed in one step through cycloaddition reactions of those Pd-π-allyl zwitterions under asymmetric catalysis,which is usually difficult to obtain with other methods.
The discovery of new Pd-π-allyl zwitterions has enabled the construction of the diverse set of carbon- and heterocyclic compounds and improves the high potential of such transformation for the synthesis of highly complex organic molecules. However,the design and exploration of unprecedented dipolar cycloadditions is particularly challenging because these dipole intermediates are often highly reactive and short-lived,leading to multiple unexpected reaction pathways. This paper reviews representative advances in Pd-π-allyl zwitterions in organic synthesis over the past five years in two aspects: (1) different kinds of new Pd-π-allyl zwitterions and their applications as different kinds of synthons in cycloaddition reactions; (2) recent advances in the early zwitterions that were hardly applied after their initial discovery. Although several related reviews have been reported [20,41-44],there is no comprehensive summary for the recent development of Pd-π-allyl zwitterions. This review will provide a systematic and overall summary of the recent developments of Pd-π-allyl zwitterions,including three different types of Pd-stabilized zwitterions (bearing a carbanion,O-centered anion or N-centered anion),as well as the related substrate applicability and reaction mechanisms.
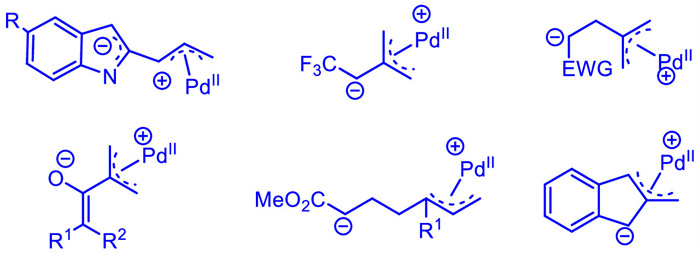
A wide assortment of compounds,including γ-methylene-δ-pentyl lactones,vinylcyclopropanes,and silylated allylic substrates were commonly used to generate Pd-zwitterionic species with a carbanion. In the past years,several novel Pd-π-allyl zwitterions were developed by using new precursors (Scheme 2). They can serve as C3,C4,and C6 synthons to participate in cycloaddition reaction with various dipolarophiles.
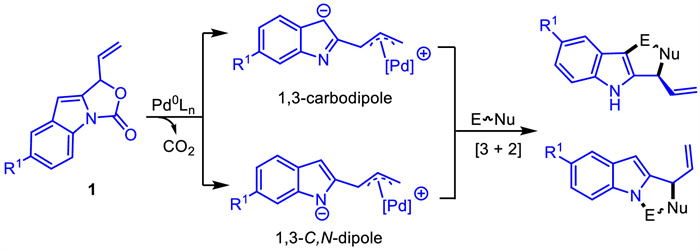
In 2020,vinyl indoloxazolidones 1 were reported as either 1,3-C,N-dipole or 1,3-carbodipole precursors in Pd-catalyzed [3 + 2] cycloaddition reactions by Shi group [45] and Deng group [46],respectively (Scheme 3). The indolyl substituted Pd-π-allyl zwitterionic intermediates were derived from the palladium catalyzed decarboxylation reaction of vinyl indoloxazolidones 1,which could serve as both all-carbon 1,3-dipoles and aza-1,3-dipoles through the anionic delocalization,providing a new platform for the divergent synthesis of polycyclic indoles. The reactions in which they participate as all-carbon 1,3-dipoles are presented firstly,and the reactions in which vinyl indoloxazolidones act as 1,3-C,N-dipoles will be presented later (vide infra).
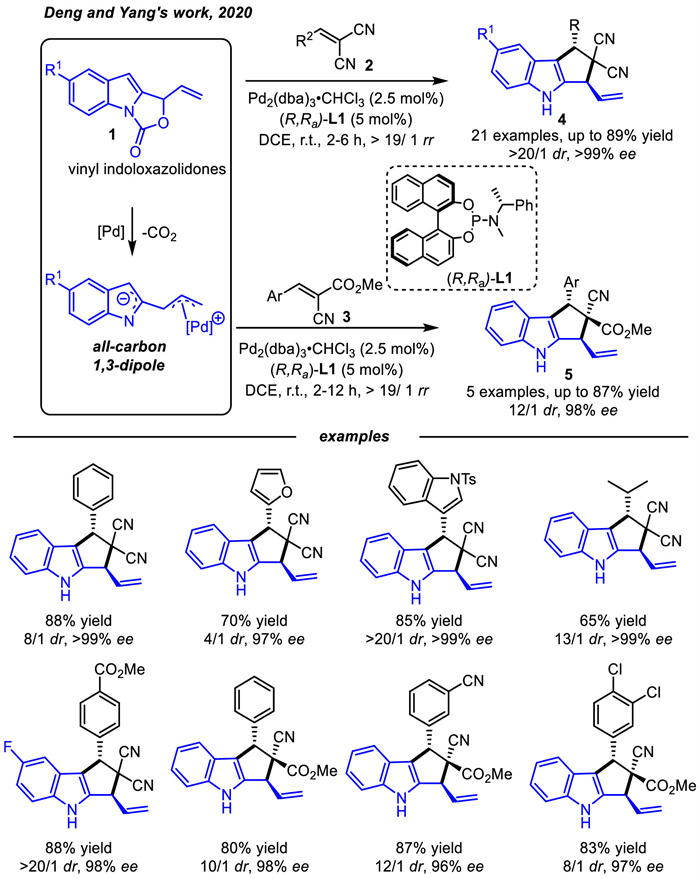
Deng and coworkers [46] first applied vinyl indoloxazolidones 1 that selectively served as all-carbon 1,3-dipole precursors in the asymmetric [3 + 2] cycloaddition with electron deficient alkenes 2 or 3,furnishing polysubstituted cyclopenta[b]indoles 4 and 5 with high regio- and stereo-selectivities in the presence of phosphoramidite ligand L1 (Scheme 4).
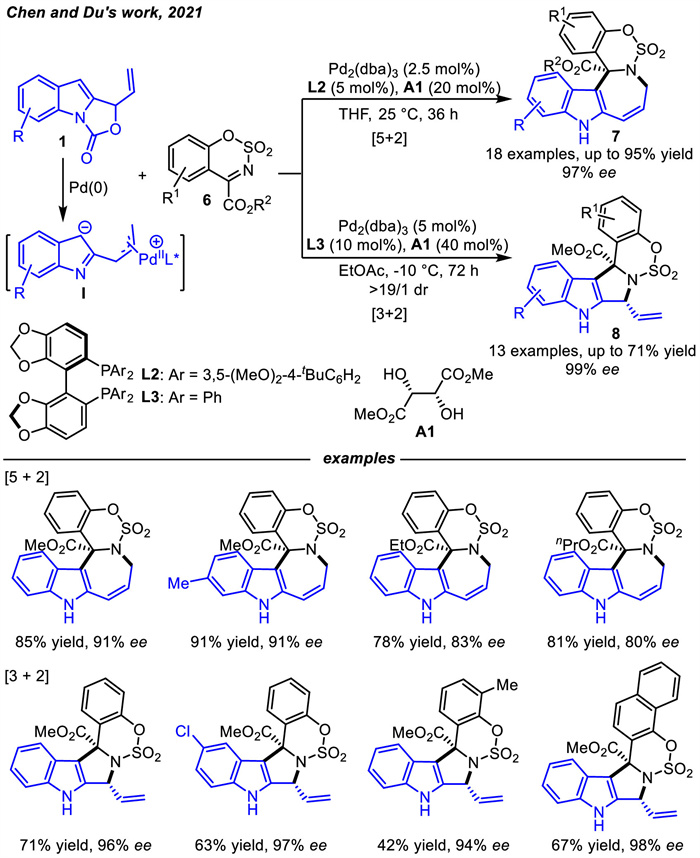
The next year,Chen and coworkers [47] also disclosed a palladium-catalyzed regiodivergent asymmetric cycloadditions of vinyl indoloxazolidones 1,which could serve as unusual 1,5-carbodipole or 1,3-carbodipole precursors by tuning chiral bisphosphine ligands and reaction conditions,in the assemblies with sulfamate-derived cyclic imines 6 (Scheme 5). A diversity of [5 + 2] cycloaddition products 7 were generally constructed with up to 95% yields and 97% enantioselectivities by employing the combination of Pd2(dba)3 and chiral bisphosphine ligand L2. Remarkably,by tuning the structure of bisphosphine ligand from a sterically bulky L2 to a less sterically demanding L3,the asymmetric [3 + 2] cycloaddition products 8 between the same substrate assemblies could be separated as the major ones in excellent stereoselectivity since the congested feature of the π-allyl palladium complex would be beneficial for the [3 + 2] cycloaddition pathway. During this process,the chiral tartrate A1 was found to be beneficial for the enantioselectivity and reactivity,probably acting as a hydrogen donor co-catalyst after assembly with the palladium(Ⅱ) intermediate via coordination. Moreover,this delocalized Pd-π-allyl zwitterions Ⅰ are similar to the zwitterionic species formed from vinylethylene carbonates (VECs) or vinyloxiranes [48-53] under Pd catalysis,which could perform as either 5-atom synthons or 3-atom synthons for cycloaddition reactions.
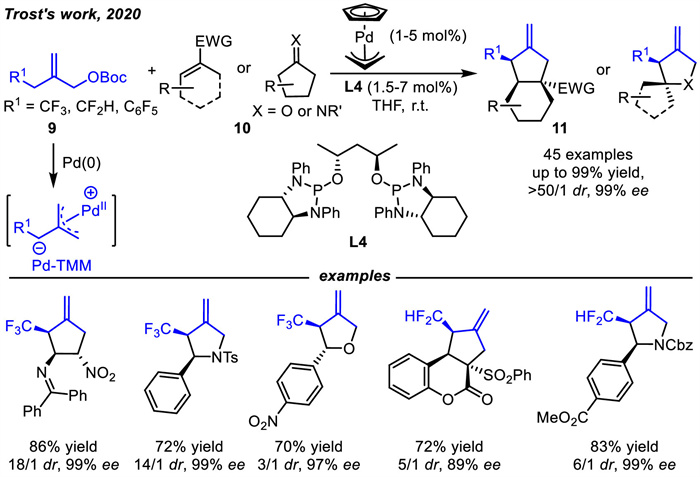
Since the seminal study by Trost and Can in 1979 [16],five-membered ring structures,such as cyclopentanes,pyrrolidines and tetrahydrofurans,have been well constructed through asymmetric [3 + 2] cyclizations of Pd-TMM with 2π-dipolarophiles. In 2020,Trost and coworkers developed a Pd-TMM containing an α-trifluoromethyl carbanion (Scheme 6) [54]. The unique charge distribution of the cationic palladium π-allyl motif may stabilize the adjacent fluorocarbanion and prevent the elimination of fluoride. Thus,asymmetric [3 + 2] cycloadditions of Pd-TMM intermediate with a vast array of acceptors 10 were realized to deliver valuable CF3-substituted five-membered rings 11 including cyclopentanes,pyrrolidines and tetrahydrofurans in high yields and stereoselectivities. In addition,other five-membered rings containing polyfluorinated substituents such as difluoromethyl,were also successfully provided using this approach.
In 2007,γ-methylidene-δ-valerolactones were first designed and used as precursors of 1,4-zwitterionic specie by Shintani and Hayashi group [17]. Since then,this type of precursors has been rapidly developed by reacting with many acceptors as an all-carbon four-atom synthon for [4 + n] cycloadditions [55-59]. Despite its great reactivity,γ-methylidene-δ-valerolactones require an electron-rich or neutral aromatic ring at the α-position of its ester group to enable the key palladium-mediated decarboxylation,which would limit the further functionalization and applications of the products. Thus,the need of more general dipole precursors and the possibility of achieving high enantioinduction have prompted chemists to explore and develop new atom-economic pathways to construct functionalized carbo- and heterocycles.
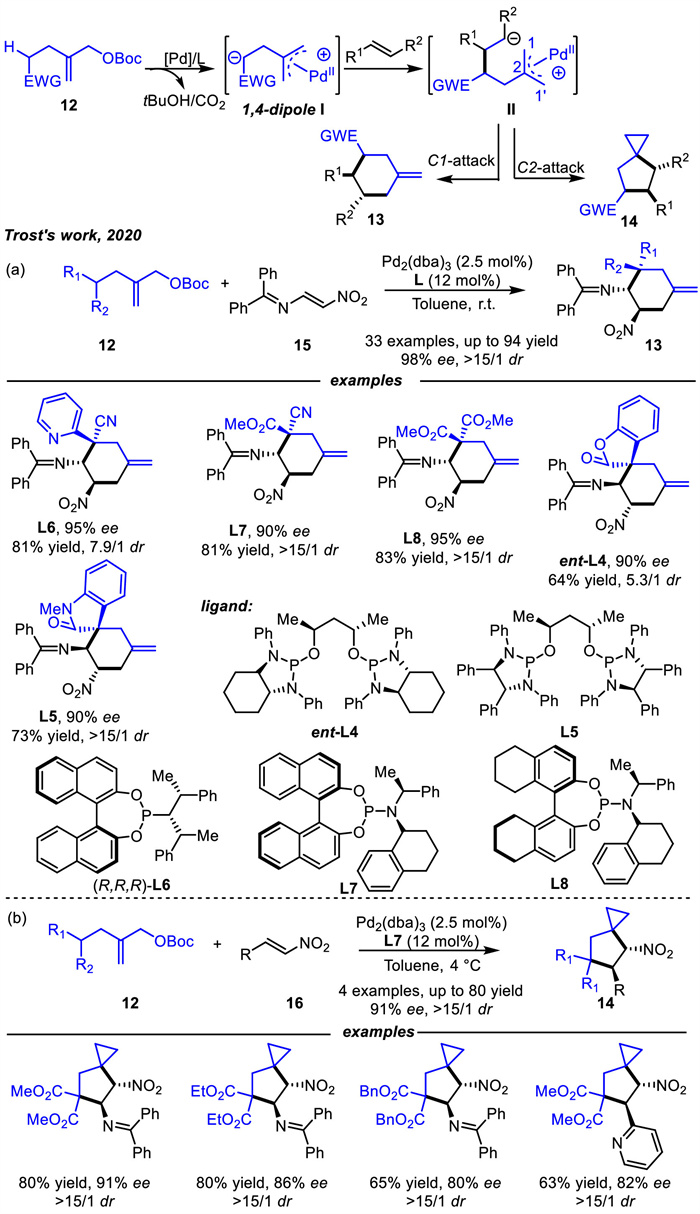
Until 2020,Trost and coworkers designed and applied a new 1,4-zwitterionic intermediate precursor that could be accessible in one step,greatly simplifying the synthesis (Scheme 7) [60]. The allyl carbonates 12 would undergo ionization in the presence of Pd(0) complex and subsequently abstract the homoallylic proton,acidified by the electron withdrawing group,with release of tBuOH and CO2. The corresponding Pd-stabilized zwitterion Ⅰ was generated,which can then be add to electron-deficient olefins 15 or 16 to deliver intermediate Ⅱ. While the subsequent ring closure of intermediate Ⅱ can yield different products because both C1 and C2 centers are electrophilic. Nucleophilic attack at the C1 terminus via a classic Tsuji-Trost type mechanism afforded 6-membered adducts 13 in up to 94% yield,98% ee and > 15/1 dr (Scheme 7a),while attack at the C2 position yielded a palladacyclobutane intermediate which subsequently would undergo reductive elimination to form the spirocyclic compounds 14 (Scheme 7b). Various chiral 6-membered rings and spiro[2.4]heptanes were prepared in high yields and selectivity through ligand-controlled and substrate induced regio- and diastereoselectivity.
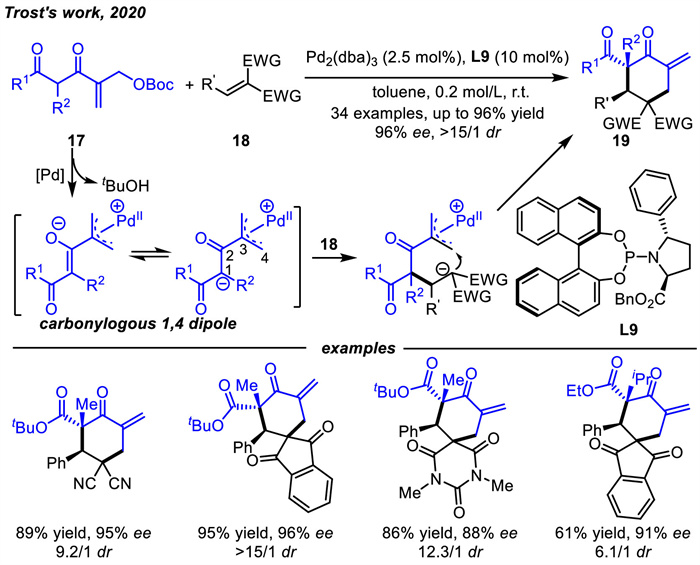
In the same year,Trost and coworkers [61] also developed another novel palladium-mediated carbonylogous 1,4-dipole by in situ deprotonation from 17 (Scheme 8). In contrast to the well-developed palladium-mediated dipole precursors that usually used electron-neutral or rich olefins,such as cinnamyl,methylmethylene. In this process,electron-deficient system was reported as the precursor. By using their developed C2-unsymmetric phosphoramidite ligand L9,this novel dipole could be used for asymmetric [4 + 2] cycloaddition reactions with various electron-deficient alkenes 18 to produce chiral cyclohexanones 19 in good to excellent results.
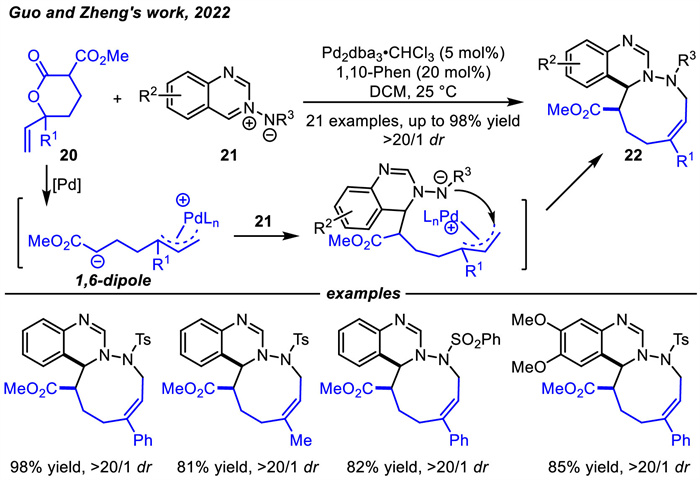
In 2022,a new precursor of π-allylpalladium zwitterion,δ-vinylvalerolactone 20,was designed for the [6 + 3] decarboxylative cycloaddition reactions with azomethine imines 21 by Guo and coworkers (Scheme 9) [62]. A range of nine-membered 1,2-dinitrogen-containing heterocycles 22 were synthesized in 77%–98% yields with > 20/1 dr. δ-Vinylvalerolactones 20 with aryl,2-naphthyl,thienyl and methyl-substituted all displayed well reactivity,affording the corresponding products.
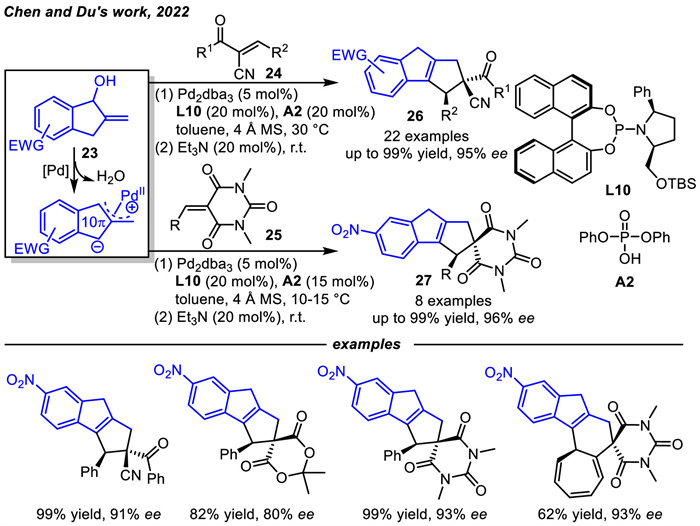
In 2022,Chen and coworkers designed novel π-allylpalladium dipolar 10π-cycloaddends derived from 2-methylene-1-indanols 23 via oxidative addition under Pd(0) catalysis and deprotonation (Scheme 10) [63]. Thus,an asymmetric [10 + 2] cycloaddition reaction of 23 with diverse activated alkenes was developed under double activation of Pd(0) and phosphoric acid. By employing a newly designed chiral phosphoramidite ligand L10,various polycyclic frameworks embedding an indene core were obtained in moderate to excellent yields with high enantioselectivities. Notably,introducing an electron-withdrawing group at the indane ring is required to enhance the acidity of the benzylic C-H. The addition of phosphoric acid as a co-catalyst may be beneficial for the oxidative addition of Pd(0) to allyl alcohol and may also facilitate enantiomeric control. Besides,apart from α-cyano chalcones 24 and barbiturate-derived alkenes 25,other types of activated alkenes can also be employed to construct polycyclic frameworks with more structural diversity through asymmetric [10 + 8] and [10 + 4] cycloaddition reactions.
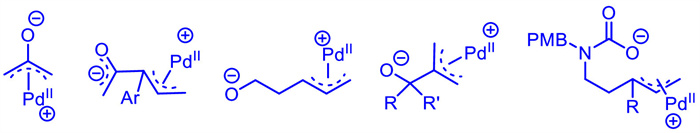
Novel Pd-π-allyl zwitterions bearing an O-centered anion have also been rapidly developed and can be used as versatile multi-atomic synthons depending on the reaction conditions and the variation of dipolarophiles (Scheme 11),however,the control of the reaction site as well as the stereoselectivity is difficult. Nevertheless,with the continuous efforts of chemists,numerous carbon- or heterocycles compounds have been successfully furnished.
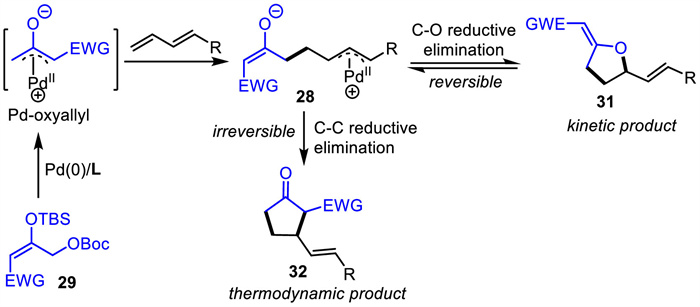
Pd-oxyallyl intermediates have attracted the attention of researchers since the 1990s [64-66]. They were generated from different precursors and were all found to react only with norbornene type strained alkenes to yield unexpected cyclopropanation products. However,it was not until very recently that a catalytic Pd-oxyallyl-mediated [3 + 2] cycloaddition was achieved by Trost and coworkers in 2018 [67]. The key to success was the introduction of an additional electron-withdrawing group (EWG) on the Pd-oxyallyl intermediate,which could drive the equilibrium toward [3 + 2] cycloaddition products (Scheme 12). The Pd-oxyallyl intermediated in situ generated by the reaction of Pd(0) with precursor 29 acted as an electrophile,which differed from the analogous Pd-TMM intermediates that are nucleophiles. Subsequently,the new Pd-allyl zwitterionic intermediate 28 would be generated by the addition of Pd-oxyallyl to the conjugated diene,and this Pd-allyl transfer process would be followed by C-O or C-C reductive elimination to form the five-membered rings 31 or 32,respectively. In addition,it was found that the kinetic products 31 would be converted to thermodynamic products 32 via the intermediate 28.
In this work,a tailored bifunctional precursor 29 and 1,3-dienes 30 including cyclohexadienes and linear dienes were chosen as substrates (Scheme 13). A wide variety of tetrahydrofurans 31 containing an exocyclic double bond were delivered in good to high yields,which was quite different from those obtained via the [3 + 2] cycloadditions of vinyl three-membered cycles with olefins [68-71].
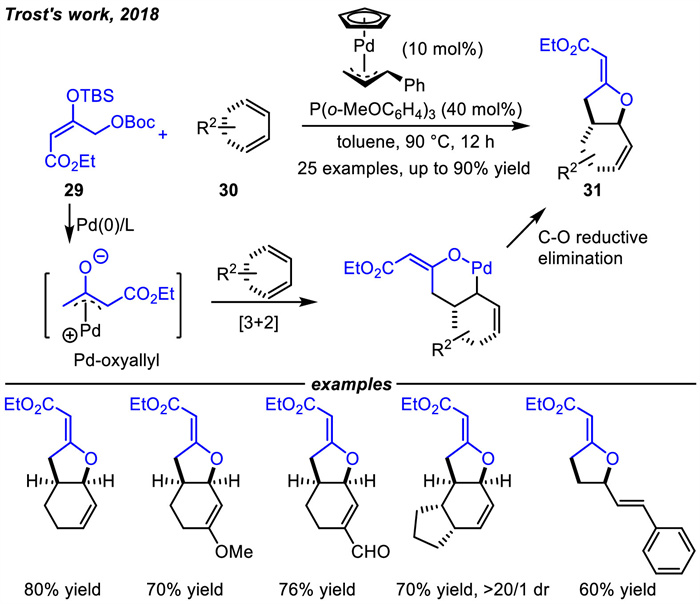
In addition,the heterocycles 31 could smoothly be converted to the thermodynamically more stable carbocyclic cyclopentanones 32 in 62%-73% yields by using CpPd(cinnamyl)/1,2-bis(diphenylphosphino)ethane (dppe) as the catalyst under elevated temperature that can facilitate the irreversible C–C bond reductive elimination (Scheme 14). Subsequently,to rationalize the regioselectivity of these reactions,Houk et al. performed density functional theory (DFT) calculations,which revealed that the C-O reductive elimination step is kinetically favorable [72]. The DFT calculations also revealed that the electron-withdrawing ester substituent was crucial as it could decrease the LUMO energy of the Pd-oxyallyl species,leading to a more favorable energy match with the HOMO of the dienes.
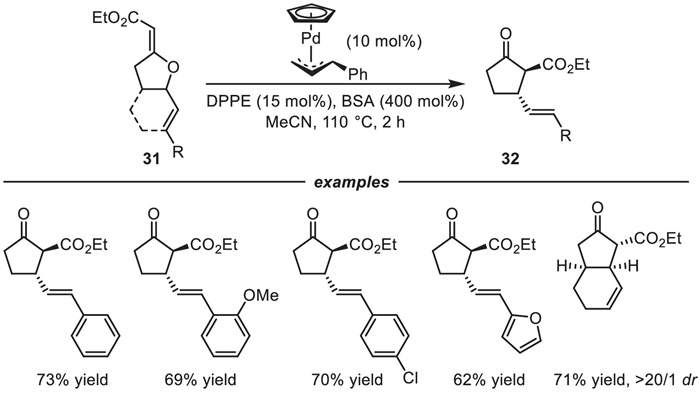
Despite the aforementioned successes,because C-O bond formation is kinetically favored,the [3 + 2] cycloadditions directly involving C-O reductive elimination are more general. In contrast,catalytic cycloaddition reactions of Pd-oxyallyl species directly through C-C reductive elimination to provide cyclic ketones remain challenging. In 2021,Zi and coworkers developed a method of lithium triflate-promoted [3 + 2] cycloaddition reactions of Pd-oxyallyl intermediates with 1,3-dienes (Scheme 15) [73]. The key to the success of this method was the coordination of the lithium ion with the alkoxide moiety,and this coordination disrupted the C-O reductive elimination pathway and promoted to form the metalenolate tethered Pd-π-allyl intermediate Ⅱ that then could undergo intramolecular allylic attack by the enolate moiety to form the carbocyclic products 35 or 36. Methylene ethylene carbonates (MECs) 34 were chosen as the precursors for the Pd-oxoallyl intermediate because they can be easily prepared by a one-step gold-catalyzed cyclization reaction [74]. The corresponding five-membered carbocycles 35 were delivered in good to high yields with high regioselectivities through [3 + 2] cycloadditions. Furthermore,the competitive [4 + 3] cycloaddition reactions were also accomplished by tuning the steric properties of the ligands. A larger sterically hindered ligand L12 facilitated delivery of [4 + 3] products 36. Importantly,DFT calculations indicated that when L12 was used as the ligand,the activation energies of the [3 + 2] and [4 + 3] cycloadditions differed in the intramolecular allylic substitution step. The former is higher than the latter. Therefore,the [4 + 3] cycloaddition reaction was favored over the [3 + 2] cycloaddition,which was consistent with experimental observations.
In 2022,Zhang and coworkers developed asymmetric [3 + 2] cycloaddition reactions of Pd-oxyallyl specie precursors 29 or 34 with cyclic or acyclic 1,3-dienes to provide various tetrahydrofuran rings in high yields and selectivities,with 10 examples in yields up to 91% and ee values up to 98% [75]. In this process,a rationally designed chiral sulfinamide phosphine (Sadphos) type ligand was employed to improve the reaction efficiency and enantioselectivity.
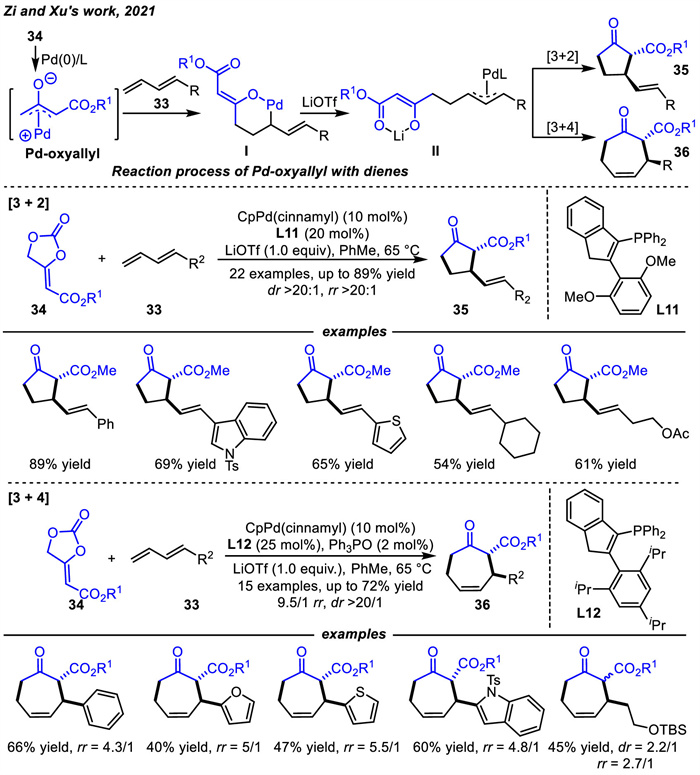
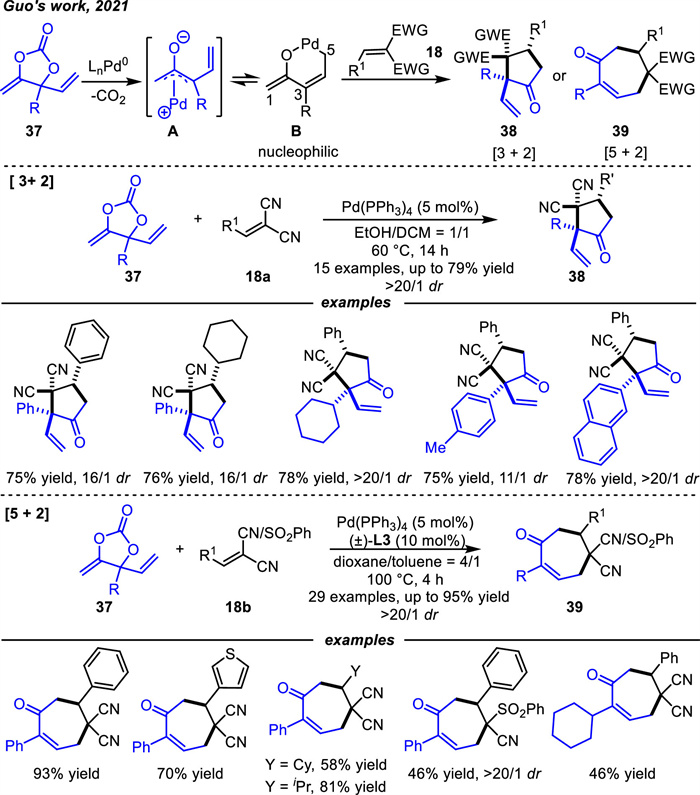
With the development of heterocycles decarboxylative process,Guo's or Zi's groups respectively designed new cyclic carbonates vinyl methylene cyclic carbonates (VMCCs) that can provide carbon-carbon zwitterion species under palladium catalysis in 2021 [76,77].
Guo and coworkers [76] designed a series of vinyl methylene cyclic carbonates 37 as substrates that could be used as 1,3- or 1,5-zwitterion intermediates under palladium catalysis (Scheme 16). The general process of the reaction is briefly described. The VMCCs could react with Pd(0) to form vinyl-substituted Pd-oxyallyl species A with the loss of one molecular CO2. The charge delocalization led to the equilibrium between A and Pd-oxypentadienyl species B. The C1 position of B had some nucleophilicity,allowing for the realization of an inverse electron demanding cycloaddition,which was quiet unlike the above electrophilicity of Pd-oxoallyl intermediate shown in Scheme 12. The five-membered carbocycles 38 were delivered through a [3 + 2] cycloaddition reaction in the presence of Pd(PPh3)4 catalyst with good to high yields and high diastereoselectivity and regioselectivity at 60 ℃. Interestingly,by adding the ligand L3 and increasing the temperature,the regioselectivity was completely reversed to [5 + 2] cycloadditon,yielding the seven-membered products 39. In addition,in order to broaden the applicability of this reaction,the asymmetric examples were also explored,but the results were not satisfactory.
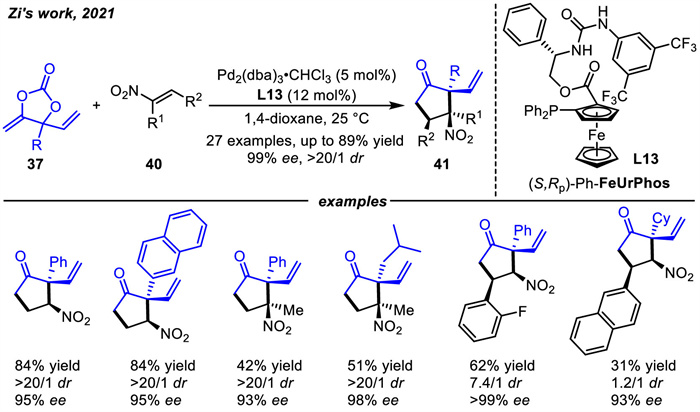
Almost at the same time,Zi and coworkers [77] realized an asymmetric [3 + 2] cycloaddition reaction of VMCCs 37 as precursors of vinyl-oxyallyl-Pd species (Scheme 17). In this case,Pd-oxoallyl was predicted by DFT calculations to be a highly electrophilic intermediate,and previously only electron-rich olefins could be available as cycloaddition partners. In contrast,the vinyl-oxoallyl-Pd species exhibited nucleophilicity,and its application would expand the range of olefins. Thus,the first enantioselective [3 + 2] cycloaddition reactions of vinyl-oxoallyl-Pd with electron-deficient nitroalkenes 40 were realized. Different ligands,including Binap,iPr-PhOX,Phosferrox,and DACH-PhTrost ligand,failed to enable the desired [3 + 2] cycloaddition. Only using phosphoramidite-type ligands can provide the cycloaddition product 41 in 40% yield with > 20:1 dr and 40% ee. Inspired by the application of noncovalent interactions in asymmetric transition-metal catalysis [78-80],a new type of hydrogen-bond-donating phosphine ligand L13 containing a tethered urea moiety was designed. Ligand L13 can form hydrogen bonds with the electron-rich oxygen of Pd-oxyallyl,and this interaction has the potential to enhance chiral induction. Cyclopentanones 41 containing three contiguous stereocerters with ee values up to 99% and dr > 20/1 were obtained by employing rationally designed FeUrPhos L13. The R group of 37 could be aryl,benzyl,alkyl,terminal alkenyl. In addition,not only various β-aryl nitroethylenes 40 but also α-methyl nitroethylenes 40 were well employed.
Recently,Liu and coworkers realized another [3 + 2] cycloaddition reaction of VMCCs 37 with [60]fullerenes in the presence of Pd(PPh3)4 [81]. In this process,VMCCs also participated in the cycloadditions as 1,3-C,C-zwitterion intermediates,which further demonstrated the reaction versatility and broad substrate applicability of VMCCs 37 as dipolar precursors.
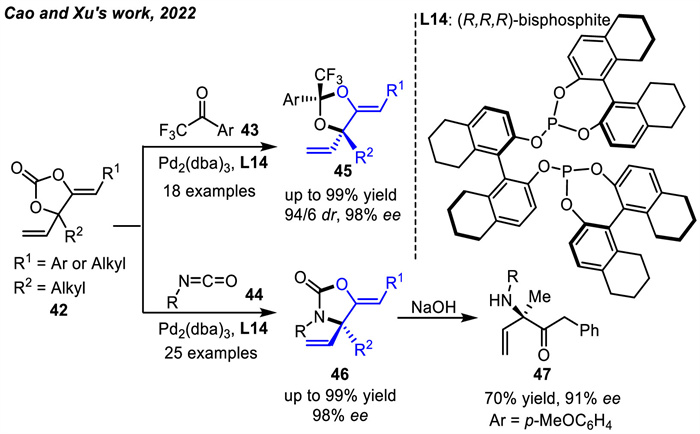
In 2022,inspired by the above works,by using chiral H8-BINOL-derived bis-phosphite ligand L14,Cao and coworkers [82] developed Pd-catalyzed enantioselective [3 + 2] cycloaddition reactions of vinyl-substituted oxyallyl carbonates 42 with activated carbonyl compounds trifluoromethyl aryl ketones 43 or isocyanates 44,delivering 1,3-dioxolanes 45 with up to 98% ee and 94/6 dr value or oxazolidinones 46 with up to 98% ee,respectively. In addition,an aminoketone 47 with α-chiral tetrasubstituted carbon was produced through ring-opening hydrolysis of 46 under basic conditions in 70% yield and 91% ee (Scheme 18).
Since the discovery of newly designed vinyl methylene cyclic carbonates (VMCCs),Guo and coworkers has achieved several Pd-catalyzed decarboxylative cyclizations to construct carbon- or heterocycles. During the process,they unexpectedly discovered the formation of pyrrole products when the reactions proceeded in the presence of amine nucleophile reagents [83]. The synthesis of a series of polysubstituted pyrroles 49 was then achieved under Pd catalysis (Scheme 19a). This decarboxylative protocol generated only CO2 and H2O as byproducts and operated at room temperature in air. Mechanistic investigations suggested that the stereoselective formation of the (Z)-configured γ-amino ketone intermediate A was crucial for the success of the reaction. The (E)-A was stable and isolatable. In contrast,the intramolecular nucleophilic attack of (Z)-A readily occurred to give intermediate B,which was subsequently dehydrated to give the corresponding pyrrole compounds 49.
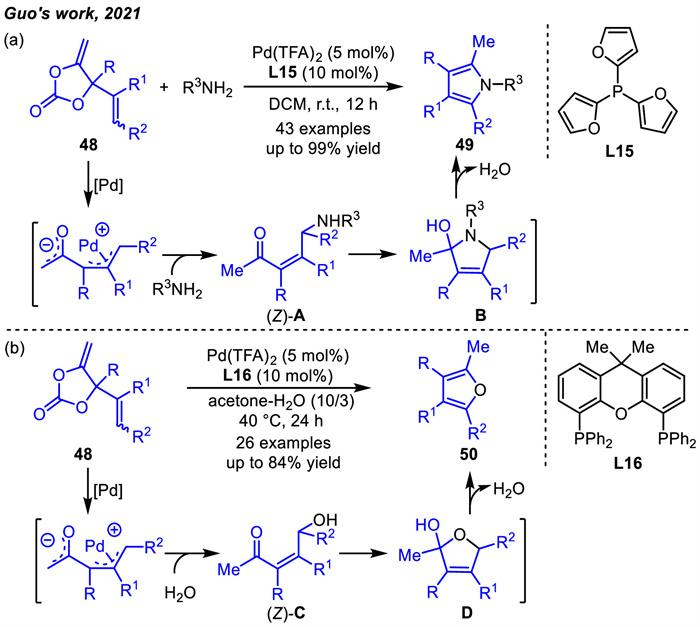
In the same year,Guo and coworkers reported a water-mediated catalytic decarboxylation process to form a series of polysubstituted furans 50 (Scheme 19b) [84]. This protocol also utilized VMCCs 48 as reaction substrates with broad functional group tolerance in the presence of Pd(TFA)2 and L16 in a mixed solvent of acetone and H2O.
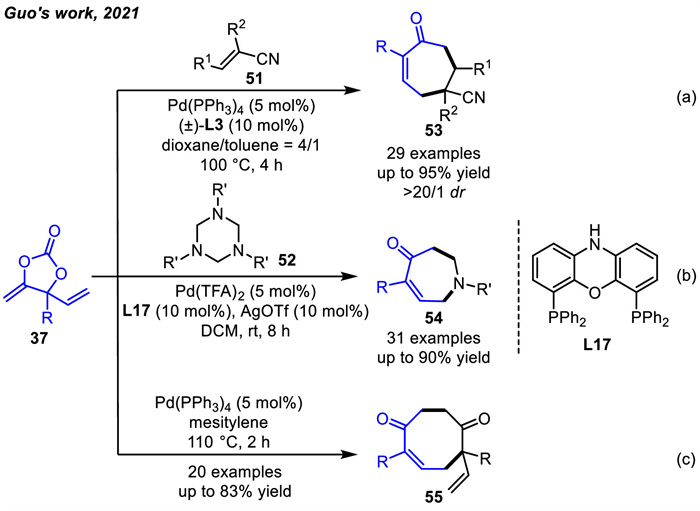
In addition to acting as three-atom synthons,VMCCs can also serve as five-atom synthons for [5+2] cycloadditions. Guo and coworkers obtained the [5 + 2] cycloaddition products 53 from the reaction of VMCCs 37 and activated alkenes 54 with high selectivity and good to high yields under Pd(PPh3)4 and ligand L3 co-catalysis (Scheme 20a) [76]. When the reaction substrates were switched from olefins 51 to triazines 52,non-fused N-aryl azepane derivatives 54 were obtained via [5 + 2] cycloaddition reactions promoted by Pd catalysis and AgOTf [85]. In this process,in order to gain insights into the reaction pathways,DFT calculations showed that the origin of the exclusive [5 + 2] rather than the [3 + 2] cycloaddition process was due to a much lower barrier for the formation of azepanes 54 (Scheme 20b). Besides,the same group [86] found that two molecules of vinyl Pd-oxyallyl species could dimerize regiospecifically to form highly functionalized nonbridged cyclooctanoids 55. This methodology demonstrated the vinyl Pd-oxyallyl species possessing both electrophilic and nucleophilic properties (Scheme 20c).
Guo and coworkers also reported ligand-controlled Pd-catalyzed decarboxylative [5 + 4] and [5 + 2] cycloadditions by utilizing VMCCs 37 as C5-synthons [87]. When using a diphosphine ligand L16,only the thermodynamically favorable [5 + 2] cycloaddition products 57 were given (Scheme 21a). The utilization of a monophosphine ligand L15 could switch the regioselectivity,prompting the challenging nine-membered compounds 58 to be the major products via the [5 + 4] cycloadditions (Scheme 21b). A reaction mechanism was proposed. First,cyclic carbonate 37 underwent decarboxylation in the presence of a palladium catalyst to give the zwitterionic Pd-π-allyl enolate intermediate Ⅰ. Followed by nucleophilic attack on dienes 56,the new zwitterionic intermediate Ⅱ was generated. The regio-divergent nucleophilic cyclization under kinetic (path a) or thermodynamic control (path b) generated the corresponding nine- or seven-membered carbocycles,respectively,with the regeneration of the palladium catalyst.
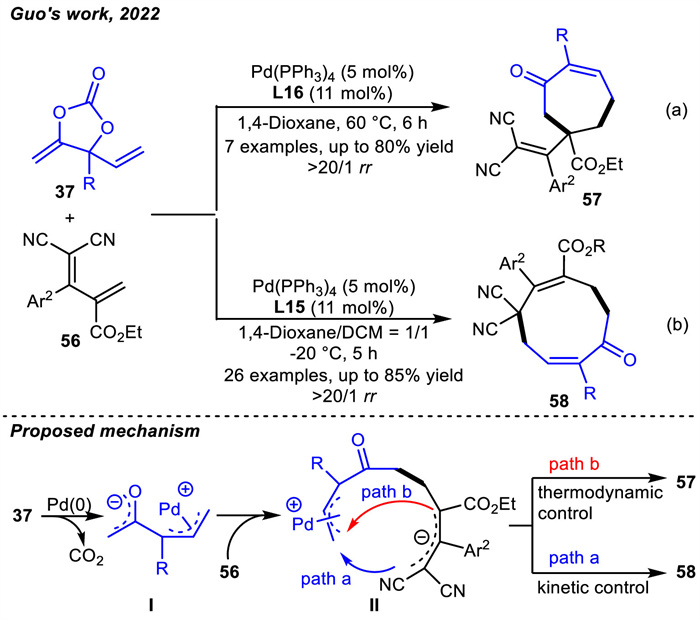
Since Alper and coworkers reported the Pd-catalyzed allylic cycloaddition of vinyloxetanes 59 with isocyanates/carbodiimides to provide compounds 1,3-oxazines in a racemic form in 1999 [88],which was the first report on the oxa-1,4-dipoles (Scheme 22). In addition to cycloaddition reactions,vinyloxetanes 59 also delivered acyclic products [89,90] and dihydropyrans [91] via ring-opening allylic substitution and ring-expansion reactions. However,metal-catalyzed asymmetric cycloaddition reactions of vinyloxetanes remained to be unknown for a long time.
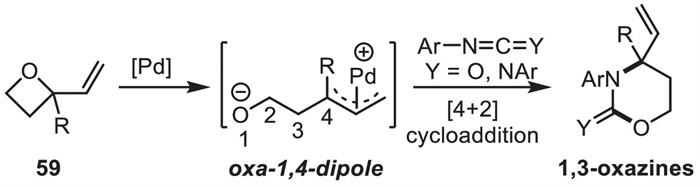
In 2019,Zhang and coworkers [92] achieved an asymmetric [4 + 2] cycloaddition of vinyloxetanes 59 with an abundant feedstock formaldehyde in the presence of Pd2(dba)3·CHCl3 and phosphoramidite ligand L18 under mild conditions to provide 4-substituted 4-vinyl-1,3-dioxanes 60 in high yields with good to excellent enantioselectivities (Scheme 23a). Vinylpropylene carbonate (VPC) 61a,which was synthesized from the corresponding 1,3-diol [93],was also tested instead of vinyl oxetanes 59 for this cycloaddition reactions. As shown in Scheme 23b,the same reaction conditions were effective for the cycloaddition of VPC 61a with formaldehyde to furnish 1,3-dioxane 60a in 60% yield with high enantioselectivity (95% ee).
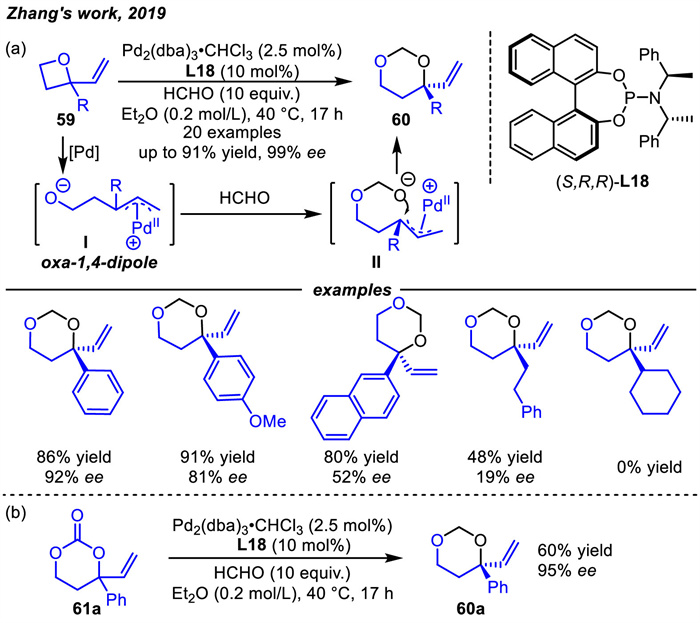
Almost at the same time,Hou and coworkers [94] paid attention to the oxa-1,4-dipoles and developed the first asymmetric [4 + 2] cycloaddition reaction of VPCs 62 with singly activated electron deficient alkenes 63 catalyzed by a combination of Pd and the newly developed benzyl-substituted P,N-ligand L19 (Scheme 24). A range of tetrahydropyrans 64 containing three continuous chiral centers were obtained in high yields with high diastereo- and enantioselectivities. Reactions using commercial P,N-ligands L20 produced 64 in high yields with varied enantioselectivities,but with very low diastereoselectivities. Excitingly,if benzylic substituted P,N-ligand (S,S)-L19 with isopropyl as a substituent at the benzylic position was used,both the yields and diastereo- and enantioselectivities were significantly improved. This class of benzylic substituted P,N-ligands was further applied in the palladium-catalyzed asymmetric [3 + 2] cycloadditions of vinyl epoxides with alkynyl esters as well [95].
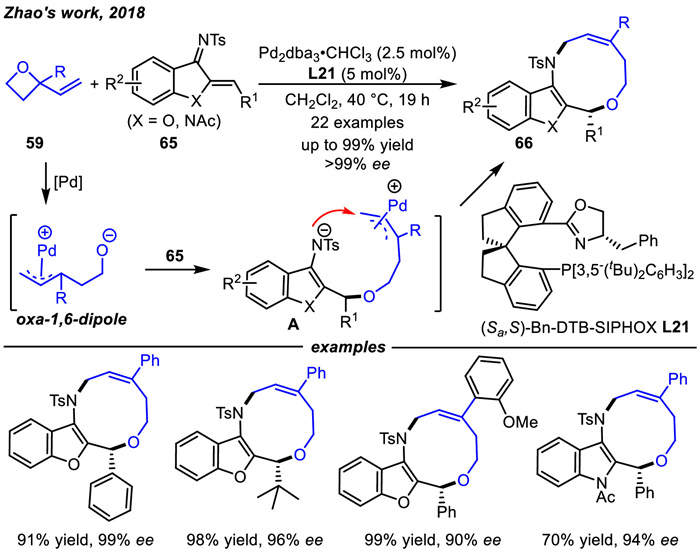
Vinyl oxetanes 59 are commonly used as 1,4-zwitterionic precursors to achieve Pd-catalyzed [4 + 2] cycloaddition reactions [96]. Until 2018,Zhao and coworkers reported the first Pd-SIPHOX L21 complex-catalyzed enantioselective [6 + 4] cycloaddition reactions of vinyl oxetanes 59 with azadienes 65 (Scheme 25) [97]. Various benzofuran- as well as indole-fused heterocycles 66 could be accessed in excellent yields and enantioselectivities. In this context,vinyl oxetanes were involved in the reaction as oxa-1,6-dipole precursors via oxidative addition of Pd(0). Then,the alkoxide moiety of oxa-1,6-dipoles underwent Michael addition with azadienes 65 to give intermediate A. Finally,the [6 + 4] cycloaddition products were obtained through intramolecular N-allylic substitution. In fact,the preparation of ten-membered rings by cycloaddition reactions remains scarce in organic synthesis. This reaction provided a fast and efficient method to construct of chiral 10-membered rings.
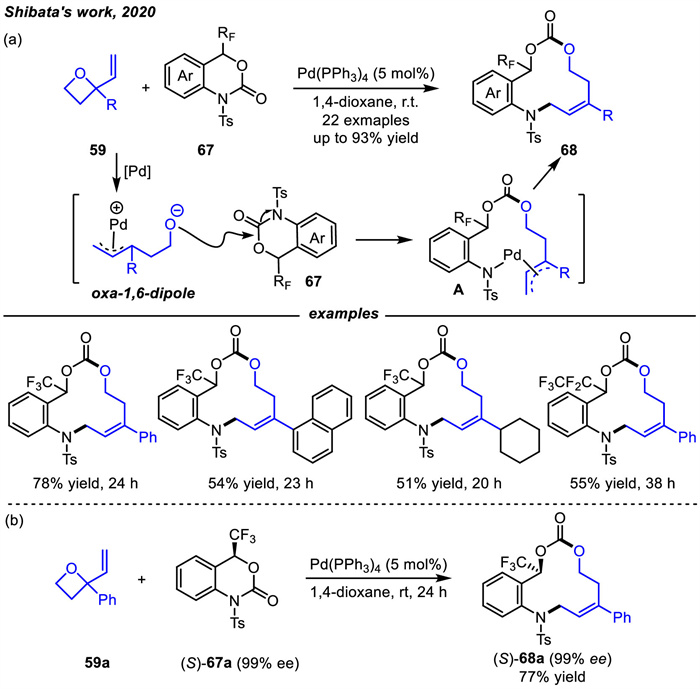
In 2020,Shibata and co-workers reported another reaction on the use of vinyl oxetanes 59 as 1,6-dipoles via Pd catalysis (Scheme 26) [98]. In this case,a non-decarboxylated Pd-catalyzed [6 + 6] cycloaddition of six-membered trifluoromethyl benzo[d][1,3]oxazinones 67 with vinyl oxetanes 59 was developed,which was an efficient way for the synthesis of trifluoromethylated 12-membered heterocycles 68. The possible reaction mechanism was shown in Scheme 26. First,the oxa-1,6-dipoles were generated in situ by the reaction of Pd with vinyl oxetanes 59. Then,nucleophilic attack on the carbonyl moiety of 67 by the oxygen anion of the oxa-1,6-dipoles induced ring opening via C-N bond breakage of 67,which led to the formation of the Pd complex intermediate A that was confirmed by LC-MS analysis (m/z = 940.0,M+Na). Finally,the reductive elimination of the Pd complex A provided [6 + 6] cycloaddition products 68. In addition,the reaction of vinyl oxetane 59a with (S)-67a under standard conditions afforded the chiral trifluoromethyl-substituted 12-membered heterocycle (S)-68a in 77% yield and 99% ee without any loss of the enantiopurity compared to the starting material (S)-67a.
In the same year,Shibata and co-workers [99] altered six-membered trifluoromethyl benzo[d][1,3]oxazinones 67 into difluorooxindoles,isatins or their analogs in reaction system,thus producing functionalized 11-membered heterocycles 70 in good to high yields by Pd-catalyzed [6 + 5] cycloadditions (Scheme 27a). In this process,vinyl oxetanes 59 smoothly in situ generated oxa-1,6-dipoles in the present of Pd2(dba)3 and dppe. The reaction mechanism was similar to the catalytic cycle of Scheme 26.
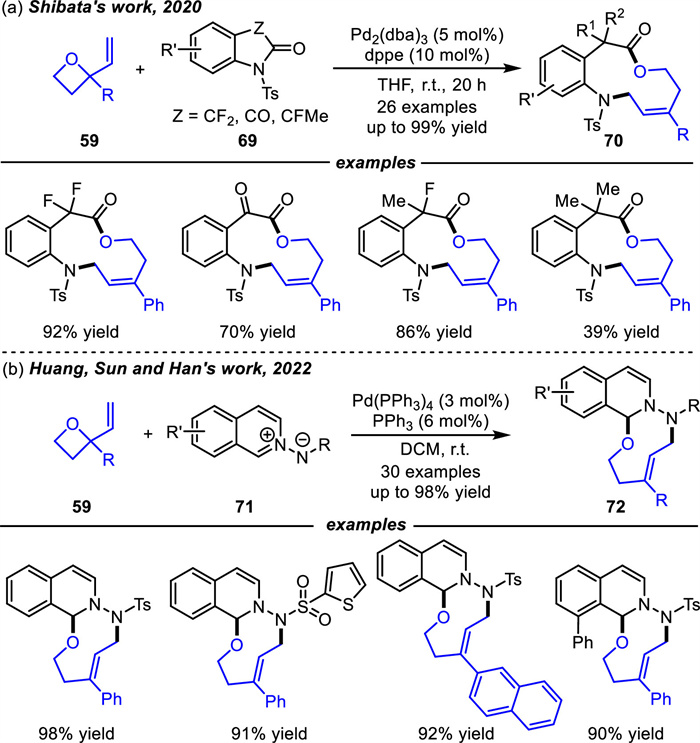
Recently,Huang and coworkers achieved a palladium-catalyzed selective [6 + 3] cycloaddition reaction of 2-vinyl oxetanes 59 and N-iminoisoquinolinium ylides 71 to deliver 9-membered N,N,O-heterocycles 72 in moderate to high yields (Scheme 27b) [100]. A range of 2-aryl-2-vinyl oxetanes 59 were examined,while 2-methyl-2-vinyl oxetanes failed to provide the corresponding heterocycle,possibly owing to the poor stability of the generated zwitterionic allylpalladium intermediates.
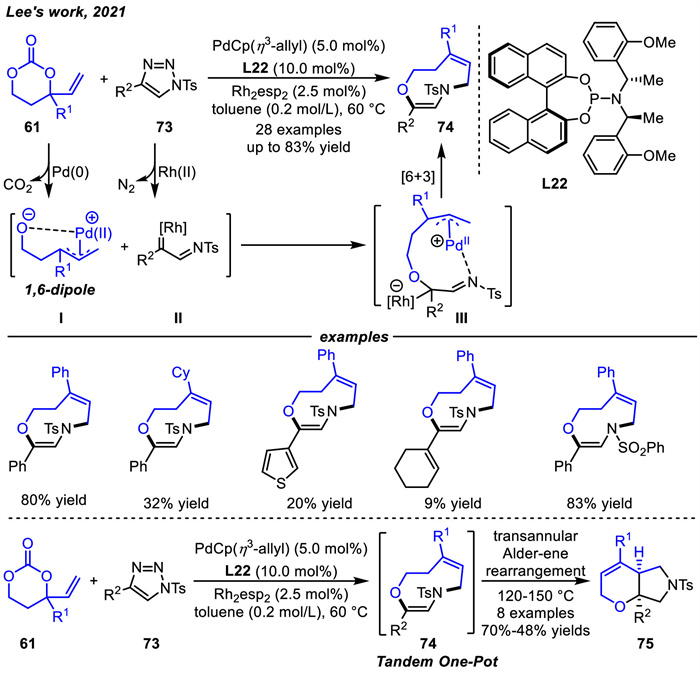
VPCs can also act as 1,6-dipole precursors,which is similar to vinyl oxetanes. VPCs could undergo oxidative addition catalyzed by palladium catalysis followed by spontaneous decarboxylation to produce oxa-1,6-dipoles. In 2021,Lee and coworkers [101] reported a novel Pd(0)/Rh(Ⅱ) dual catalytic strategy to enable [6 + 3] cycloadditions between VPCs 61 and N-sulfonyl-1,2,3-triazoles 73 to provide nine-membered 1,4-oxazonines 74 in moderate to high yields (Scheme 28). The plausible reaction pathway was depicted. First,the Pd(0) and Rh(Ⅱ) catalysts selectively activated 61 and 73,respectively,to generate 1,6-dipole Ⅰ and 1,3-dipole equivalent α-imino rhodium(Ⅱ) carbenoid intermediate Ⅱ. Then,the oxygen anion of intermediate Ⅰ could be added nucleophilically to the electrophilic carbenoid carbon of intermediate Ⅱ to deliver the Pd/Rh-bimetalated intermediate Ⅲ. Finally,nine-membered oxazonines 74 were formed through intramolecular cyclization pathways. Moreover,the nine-membered oxazonines 74 could be further converted to the corresponding cis-fused bicyclic hexahydropyranopyrroles 75 in moderate to good yields through the transannular Alder-ene rearrangement in one pot.
In addition to VPCs,2-alkylidenetrimethylene carbonates (ADTMCs),first reported by Tsuji and coworkers [102] in 1984,were also applied to Pd-catalyzed cycloaddition reactions with isocyanates by Hayashi and co-workers in 2011 (Scheme 29) [103]. The application of ADTMCs has developed rapidly in recent years. Interestingly,compared to VPCs or vinyl oxetanes,ADTMCs were used only as 1,4-dipole species for Pd-catalyzed intermolecular cycloadditions with different dipolarophiles,and the cycloaddition products contained an exocyclic double bond.
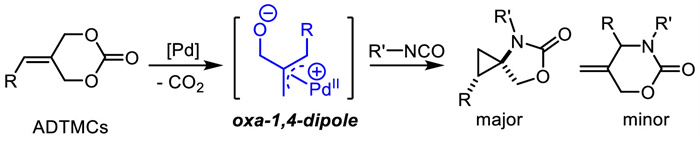
In 2019,Liu and coworkers [104] explored a Pd-catalyzed [4 + 2] cycloaddition between ADTMCs 76 and fullerene 77 to afford fullerene-fused tetrahydropyrans 78 in medium to good yields (Scheme 30). Although there were only 5 examples,this methodology revealed the application of ADTMCs as oxa-1,4-dipolar precursors under palladium catalysis.
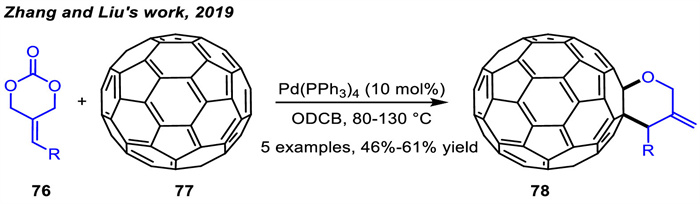
In 2020,Guo and co-workers [105] realized palladium-catalyzed asymmetric [4 + 2] cycloadditions of ADTMCs 76a with olefins derived from pyrazolones 79,indandiones 80 or barbiturates 25 to provide pharmacologically interesting chiral tetrahydropyrane-fuse spirocyclic scaffolds 81-83 in good to excellent yields and high enantioselectivities (Scheme 31). The chiral diphosphine ligand L23 was proved to be the best choice. In this process,ADTMCs were designed to generate oxa-1,4-dipoles via palladium-catalyzed oxidative addition followed by spontaneous decarboxylation.
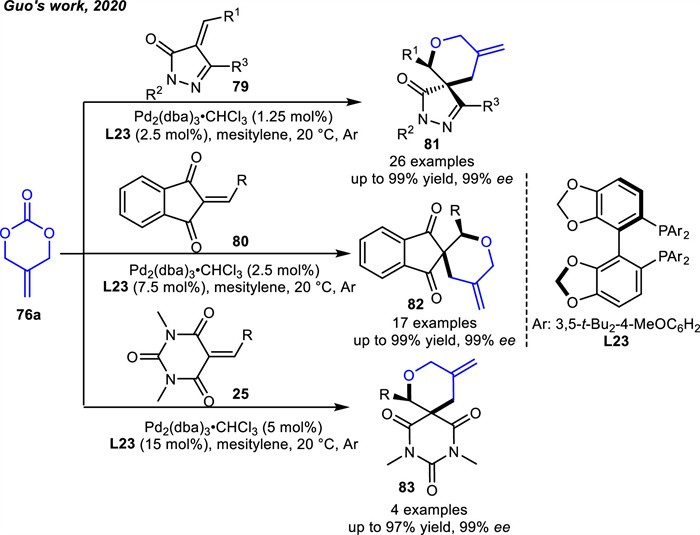
In the same year,Shibata and coworkers [99] applied 76a in the Pd-catalyzed [4 + 5] cycloaddition reactions with isatins and their analogs to access 9-membered heterocycles 84 in good to excellent yields (Scheme 32). The highly electron withdrawing nature of the Z group in combination with the nucleophilic attack of the in-situ generated 1,4-dipole species induced the C-N bond breakage of substrates 69. The desired products 84 were obtained by subsequent intramolecular cyclization.
In 2021,Lu and co-workers successfully completed the Pd-catalyzed,visible-light-induced asymmetric [4 + 2] cycloaddition reaction of ADTMCs 76 with α-adiazoketones 85 (Scheme 33) [106]. The proposed mechanism began with the generation of oxa-1,4-dipole intermediates Ⅰ from the reaction of ADTMCs and Pd(0) catalyst. Meanwhile,ketenes Ⅱ were readily generated from α-diazoketones 85 via a photo-Wolff rearrangement under blue LEDs. Then,intermolecular nucleophilic addition/intramolecular asymmetric allylic alkylation sequence produced 6-membered lactones 86 bearing exocyclic olefin. The chiral phosphoramidite ligand L24 proved to be efficient and provided chiral products 86 in 50%-95% yields with high selectivities. The reaction had a wide range of substrate applicability,allowing the use of α-diazoketones containing Me,Et,nBu,i-Bu,Bn and so on. Aryl substituted ADTMCs with different electrical properties could be employed. Moreover,unsubstituted ADTMCs also reacted smoothly; whereas alkyl-substituted ADTMCs failed to produce the desired lactone products under the standard conditions.

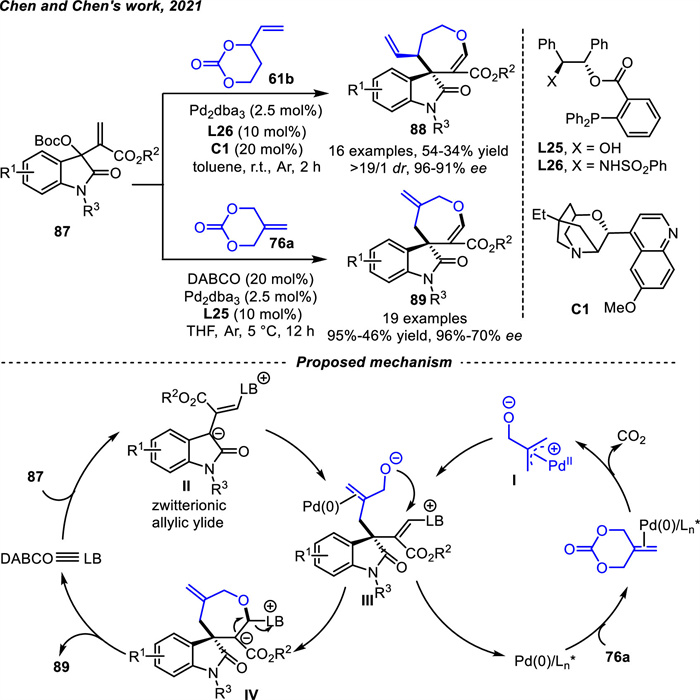
Chen and coworkers [107] achieved that the isatin-derived Morita-Baylis-Hillman (MBH) carbonates 87 could in situ form zwitterionic allylic ylides Ⅱ under the catalysis of Lewis bases (LBs),such as DABCO,DMAP or PPh3 (Scheme 34). ADTMCs 76a or VPCs 61b would readily generate zwitterionic Pd-π-allyl intermediates upon Pd-catalyzed CO2 dissociation. Therefore,an asymmetric [4 + 3] cycloaddition reaction between isatin-derived MBH carbonates 87 and oxygen-containing 1,4-dipoles through the process of intermolecular allyl substitution/intramolecular cyclization was realized (Scheme 34) [108]. A range of spirooxindoles incorporating an oxepane frameworks 88 or 89 were smoothly constructed in moderate to good yields with high stereocontrol. Phosphine ligands bearing hydrogen bonding motifs were suitable,such as L25,L26,which could be readily obtained from chiral 1,2-aminoalcohol or 1,2-diol,respectively [77,109]. In this process,the hydrogen bonding interactions played a pivotal role in enhancing reactivity and enantiocontrol.
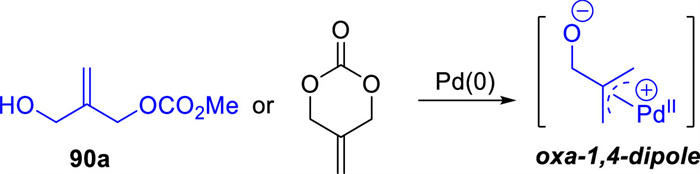
Analogous to ADTMCs,3-hydroxy-2-methylenepropyl carbonates could be transformed in situ into the same oxa-1,4-dipole intermediate under palladium catalysis (Scheme 35). The 3-hydroxy-2-methylenepropyl methyl carbonate 90a,first reported by Pátek and co-workers in 1996 [110],was synthesis in one step. The allyl carbonates bearing a nucleophilic alcohol side chain have been applied in Pd-catalyzed [4 + 2] cycloadditions with indoles 91 or para-quinone methides 93 by You's [111] or Yao's groups [112] in 2016 and 2017,respectively (Schemes 36a and b). Since then,the application of 3-hydroxy-2-methylenepropyl carbonates with a nucleophilic alcohol side chain in cycloadditon reactions has been opened.
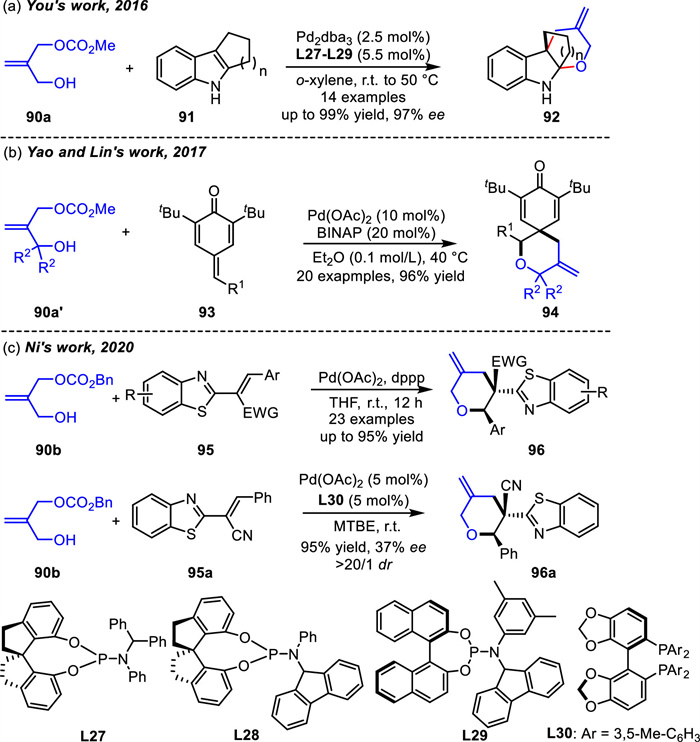
In 2020,3-hydroxy-2-methylenepropyl carbonate 90b was further used to deliver an array of 3-methylenetetrahydropyrans 96 in good to excellent yields via [4 + 2] cycloadditon reactions with 2-alkenylbenzothiazoles 95 by Ni and coworkers (Scheme 36c) [113]. When asymmetric syntheses were attempted,Trost ligands or monodentate phosphoramidite ligands were tested. Unfortunately,neither was able to yield the product of the [4 + 2] cycloaddition reaction. Chiral bidentate ligands were also tested,and only axially chiral bisphosphine ligand L30 realized the enantioselectivity and gave 96a in 95% yield and 37% ee.
Subsequently,Lin and coworkers [114] developed the ligand-controlled asymmetric [4 + 2] or [4 + 4] cycloaddition reactions of tert-butyl[2-(hydroxymethyl)allyl]carbonate 90c with benzofuran-derived azadienes 65a by palladium catalysis (Scheme 37). Using chiral P,P-ligand (S)-ClMeO-BIPHEP L31,the tetrahydropyran-fused spirocyclic compounds 97 with good to high enantio- and diastereoselectivities were obtained in good yields via the [4 + 2] cycloaddition reactions of intermediate Ⅰ (Scheme 37). When taking advantage of chiral P,N-ligand (S,Rp)-PPFA L32,the chemo- and regio-selectivities were switched to synthesize a variety of benzofuro[2,3-c][1,5] oxazocines 98 in good yields with excellent enantioselectivities via the [4+4] cycloaddition reactions of intermediate Ⅱ. The C- or N-nucleophilic sites could be specifically discriminated during the subsequent intramolecular allylic substitution processes after initial oxo-Michael addition due to the inherent steric and electronic effects of two different chiral ligands.
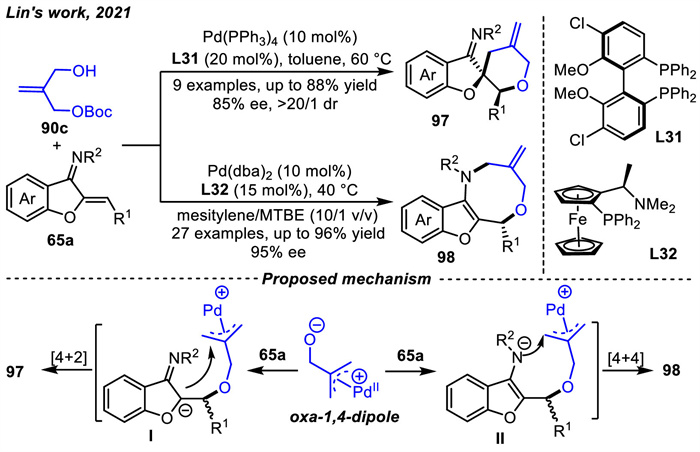
Almost simultaneously,Archambeau and coworkers [115] also realized a similar [4 + 4] cycloadditon reaction using dppe as the ligand between benzofuran-derived azadienes 65a with tert-butyl[2-(hydroxymethyl)allyl] carbonate 90c. In contrast to the above work,the difference was the absence of asymmetric examples. Noticeably,linear azadienes were also involved and yielded monocyclic eight-membered heterocycles with complete regioselectivity.
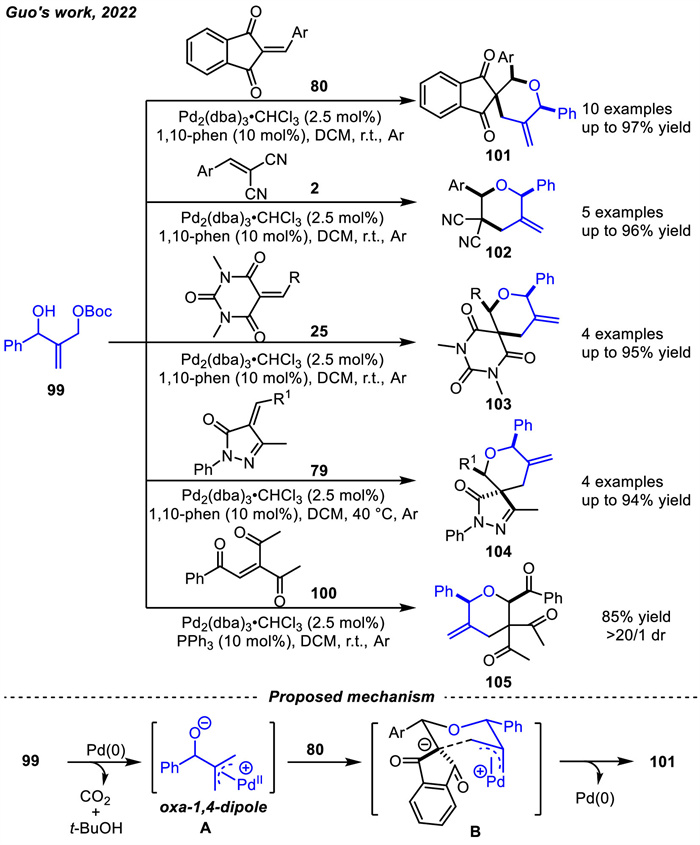
In 2022,Guo and coworkers [116] achieved the construction of tetrahydropyran derivatives 101-105 in moderate to high yields with excellent dr values ( > 20:1 dr) via palladium-catalyzed [4 + 2] cycloaddition reactions of hydroxy-tethered allyl carbonate 99 with five electron-deficient alkenes,including indandione-based alkene 80,1,1-dicyanoalkenes 2,barbiturate-derived alkenes 25,pyrazolone-derived alkenes 79,and acyl-substituted alkene 100 (Scheme 38). Interestingly,compared with the previous 3-hydroxy-2-methylenepropyl carbonates,the α-position of oxygen atom in the dipole precursors has one extra substituent in 99,and therefore the α-position of the oxygen atom in the corresponding tetrahydropyran products also has an additional chiral center. The possible mechanism is described using 99 and indandione-based alkenes 80 as substrates. First,the oxa-1,4-dipole intermediate A is produced by palladium-catalyzed decarboxylation/deprotonation of substrate 99 to release of CO2 and t-BuOH. The intermediate A subsequently attacks indandione-based alkenes 80 to give the intermediate B. The two aryl groups in the six-membered ring transition state of B are located at equatorial positions,so that the cis-isomer serves as the major product when undergoing intramolecular cyclization.
Very recently,Yuan and coworkers also developed the [4 + 2] cycloaddition reaction of 2-alkylidenetrimethylene carbonates 76 or 2-(hydroxymethyl)-3-arylallyl carbonates 90 with 3-nitroindoles to deliver a wide range of indoline-fused tetrahydropyrans in good yields with excellent diastereoselectivities [117]. That was due to the formation of the same oxa-1,4-dipole intermediate catalyzed by palladium using different dipole precursors. Unexpectedly,Liu and coworkers also involved 2-alkylidene-trimethyl carbonates (ADTMCs) as 1,3-all-carbon dipoles in the [3 + 2] cycloaddition reactions with [60]fullerenes [81].
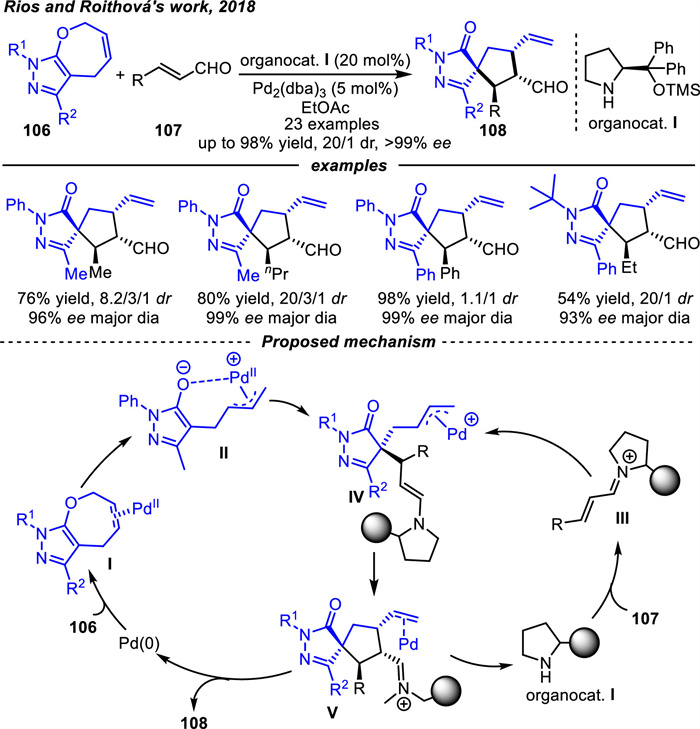
In 2018,Rios and coworkers [118] successfully reported the first ring contraction/formal [6 + 2] cycloaddition of pyrazolone derivatives 106 with α,β-unsaturated aldehydes 107 in synergistic Pd(0)/chiral secondary amine catalysis to obtain [5,5]-spiropyrazolone derivatives 108 in excellent yields and stereoselectivities (Scheme 39). The reaction mechanism started with the coordination of palladium to the double bond of substrates 106. Palladium not only interacted with the C=C double bond,but also coordinated with one molecule of acetonitrile to form complex Ⅰ. Complex Ⅰ rearranged to form intermediate Ⅱ. Meanwhile,iminium intermediate Ⅲ was formed via condensation of enal 107 with a chiral secondary amine,which underwent conjugate addition with intermediate Ⅱ to give complex Ⅳ. The desired products 108 were afforded after intramolecular allylic substitution and hydrolysis of iminium. In addition,the protonated form of the key palladium-activated intermediate Ⅱ can be detected by mass spectrometry and its structure was characterized by infrared spectroscopy and DFT calculations.
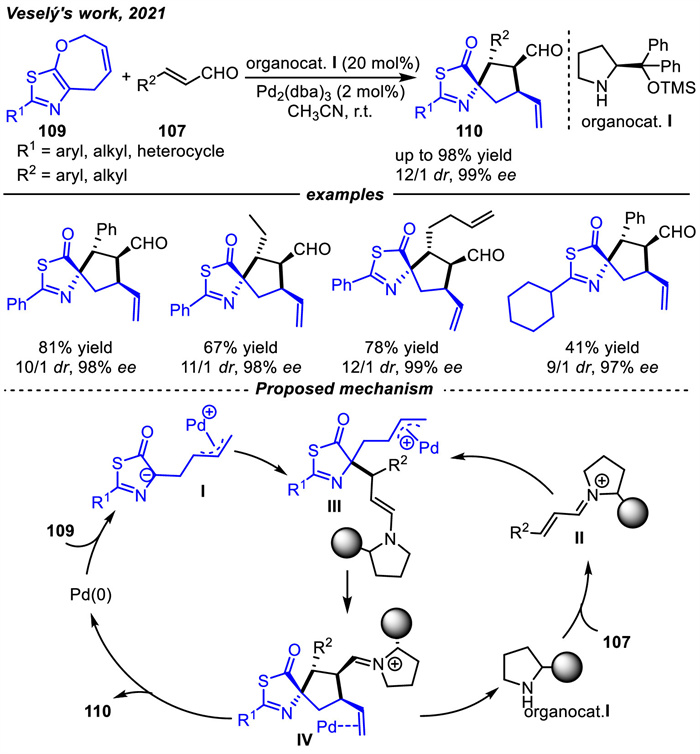
In 2021,Veselý and coworkers [119] also developed similar cycloadditions between thiazole derivatives 109 and α,β-unsaturated aldehydes 107. The reactions were catalyzed by the cooperative achiral Pd(0) complex and a chiral secondary amine to deliver the corresponding chiral spirothiazolones 110 in yields up to 98%,as well as 12/1 dr and 99% ee values (Scheme 40). The process proceeded by forming the transient zwitterionic π-allyl palladium intermediates Ⅰ and chiral α,β-unsaturated iminium ions Ⅱ. Next,iminium intermediate Ⅱ underwent conjugate addition to intermediate Ⅰ,affording enamine Ⅲ. After intramolecular 5-exo-trig cyclization and hydrolysis of iminium,spirocycles 110 were formed and Pd(0) was returned to the catalytic cycle.
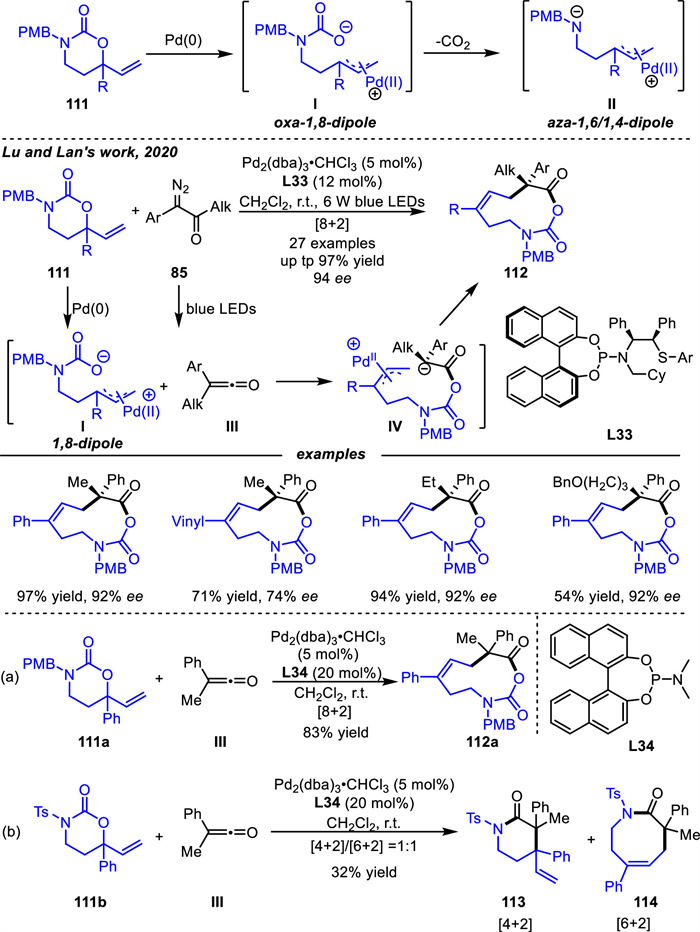
The development of 1,8-dipoles has lagged significantly compared to Pd-π-allyl 1,4- or 1,6-dipoles. In 2020,Lu and coworkers [120] developed Pd-catalyzed [8 + 2] cycloaddition reactions with α-diazoketones 85 using carbamates 111 as non-decarboxylated Pd-containing 1,8-dipoles Ⅰ rather than decarboxylated intermediate Ⅱ to provide various 10-membered monocyclic products 112 bearing chiral quaternary stereocenters in the presence of chiral ligand L33 under mild conditions (Scheme 41). Decarboxylation occurred in most dipole cycloaddition reactions based on the palladium-catalyzed reactions involving carbamate substrates,producing active Pd-π-allyl zwitterionics by releasing CO2 [44]. Interestingly,in this process,the carbonate anions of dipoles Ⅰ could attack the ketenes Ⅲ via nucleophilic addition to produce intermediates Ⅳ,and subsequently intramolecular allylation of the enolate intermediates Ⅳ would yield 10-membered monocyclic products 112. Moreover,the electronic effect of the amine played a key role in the stability of the reaction intermediates. Under the same conditions,tosyl (Ts)- and 4-MeO-benzyl (PMB)-substituted vinyl carbamates gave very different results (Scheme 41b vs. 41a). The former reaction provided an inseparable mixture of 6- and 8-membered cycloadducts 113 and 114,neither of which retained the CO2 unit (Scheme 41b). While the latter reaction did delivere the desired 10-membered monocyclic compound 112a (as racemate) in a good yield (Scheme 41a). These results are similar to that of γ-methylidene-δ-valerolactones,where the electron-rich α-substituted of γ-methylidene-δ-valerolactones could act as 1,6-dipoles by inhibiting the decarboxylative process [56].
N-Heterocycles have received widespread attention due to their bioactivities and pharmaceutical applications [121-123]. Therefore,it is necessary to develop facile and efficient dipolar cycloaddition reactions for the selective and diverse construction of various N-heterocycles. In addition to the well-known vinyl aziridines as aza-dipole precursors,in 2006,Tunge and coworkers [124] observed for the first time aza-1,4-zwitterionic Pd complexes generated by palladium catalyzed decarboxylation of 6-vinyl-1,3-oxazinanones,undergoing [4 + 2] cycloaddition with electron-deficient olefins. On the basis of those preliminary studies,several new 1,n-C,N-dipoles have been recently developed (Scheme 42).
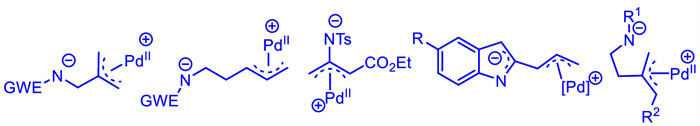
In 2016,Harrity and coworkers [125] established a short and scalable synthetic route to carbamate 115,which was able to adapt the sequence developed by Suzuki [126]. Then,the [4 + 2] cycloaddition reactions of carbamate 115 that was used as a precursor of 1,4-C,N-dipole intermediate,with 1,3-dicarbonyl substrates 116 or 118 were realized by using a catalyst system comprising Pd(dba)2 and phosphoramidite ligands (Scheme 43). When achiral phosphoramidite ligand L35 was employed,a range of piperidines 117 were constructed in good to high yields (Scheme 43b). When using chiral ligand ent-L18,increasing the steric encumbrance of R group of ketoesters 118 had varying effects on the selectivity,where the isopropyl group produced the best enantiomeric excess. The corresponding piperidines 119 were obtained in high yields and enantioselectivities (Scheme 43c).
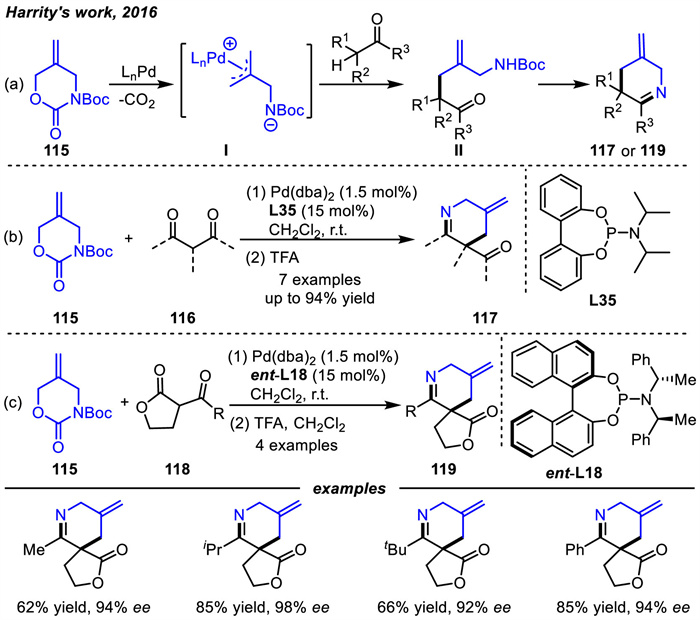
In 2021,the same group [127] achieved another [4 + 2] cycloaddition between carbamate 115 and α-fluoro β-ketoesters 120 or α-SCF3-substituted ketones 121 to construct 3-fluoro- or trifluoromethylthio-piperidines 123 and 125 with imine,ester,and alkene functional groups in good to high yields via 122 or 124 intermediates,respectively (Scheme 44). This method is a new and efficient way to introduce 3-trifluoromethylthio-group into piperidines,providing a general approach for these important scaffolds.
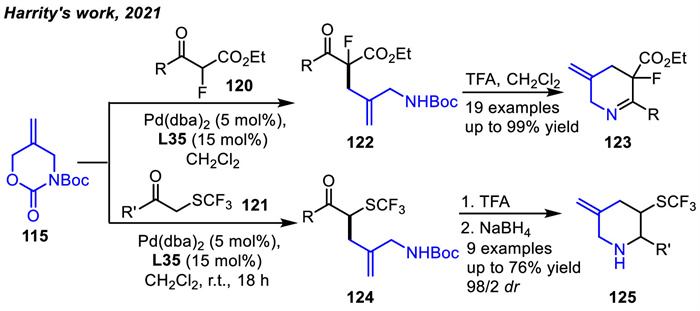
In 2019,acyclic allylic carbonates 126-128 can be readily obtained in a one-step synthesis. A Pd-catalyzed asymmetric [3 + 2] cycloaddition reaction of nitrogen-containing allylic carbonate 126 with isocyanates 44 was first reported by Zhang and coworkers (Scheme 45a) [128]. A practical and efficient method for obtaining imidazolidinones 129 in high yields and enantioselectivities in the presence of chiral binol-derived phosphoramidite ligand L18 and palladium co-catalysis was developed. Allylic carbonate 126 could afford stable 1,3-zwitterionic allylpalladium intermediate similar to 5-vinyloxazolidin-2-ones or vinylaziridine [20,129]. In addition,a Pd-catalyzed asymmetric [4 + 2] cycloaddition of nitrogen-containing allylic carbonate 127 with isocyanates 44 was also developed (Scheme 45b) [128]. Allylic carbonate 127 could provide stable 1,4-zwitterionic allylpalladium intermediate similar to 6-vinyl-1,3-oxazinanones under palladium catalysis. A series of chiral tetrahydropyrimidinones 130 could be obtained in up to 87% yield and 99% ee under ligand L36 and palladium co-catalysis. When the reaction of allyl carbonate 128 with benzyl isocyanate 44a under the same reaction conditions as that of 126 was investigated,only pyrrolidine 131 was obtained in 72% yield and 43% ee. No cycloadduct 132 was observed (Scheme 45c). This result suggested that allyl carbonate 128 underwent intramolecular cyclization to produce pyrrolidine 131 through the generation of 1,5-zwitterionic allylpalladium intermediate.
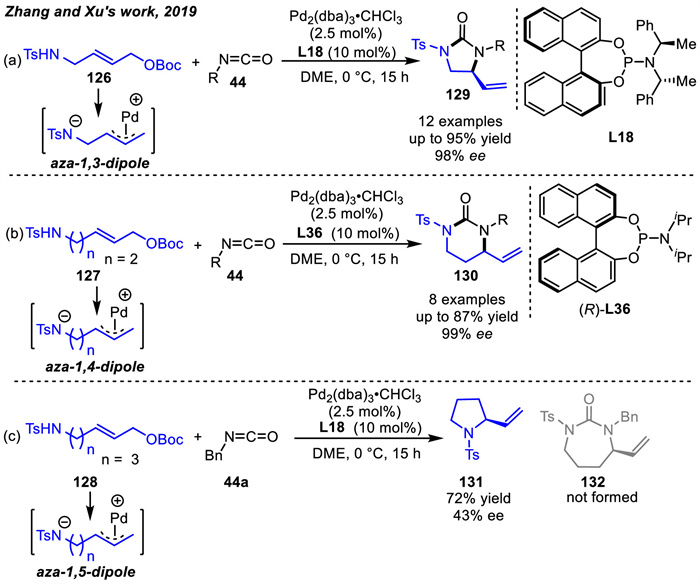
In 2019,Trost and coworkers designed a new Pd-aminoallyl precursors 133 and applied them to the [3 + 2] cycloaddition reactions with dienes (Scheme 46a) [130]. Compared with their previous reports,an electron withdrawing group on the Pd-oxyallyl precursors was essential for the smooth [3 + 2] cycloaddition with conjugated dienes [67]. Thus,the electron-withdrawing ester motif was also indispensable for the [3 + 2] cycloaddition involving the Pd-aminoallyl precursors. A variety of pyrrolidines 134 were produced in moderate to high yields via the C-N reductive elimination. The Pd-aminoallyl precursors 133 were prepared from commercially available substrates in a single step by Tamaru and coworkers in 1994 [131]. Notably,similar to Pd-oxyallyl intermediate,the heterocycle adducts 134 can be readily isomerized to thermodynamically more stable carbocycles 135 via intermediate 136 in the present of bidentate dppe ligand and CpPd(cinnamyl) (Scheme 46b). Thus,the five-membered carbocyclic 135 could be formed by palladium-catalyzed [1,3] N-to-C rearrangement.
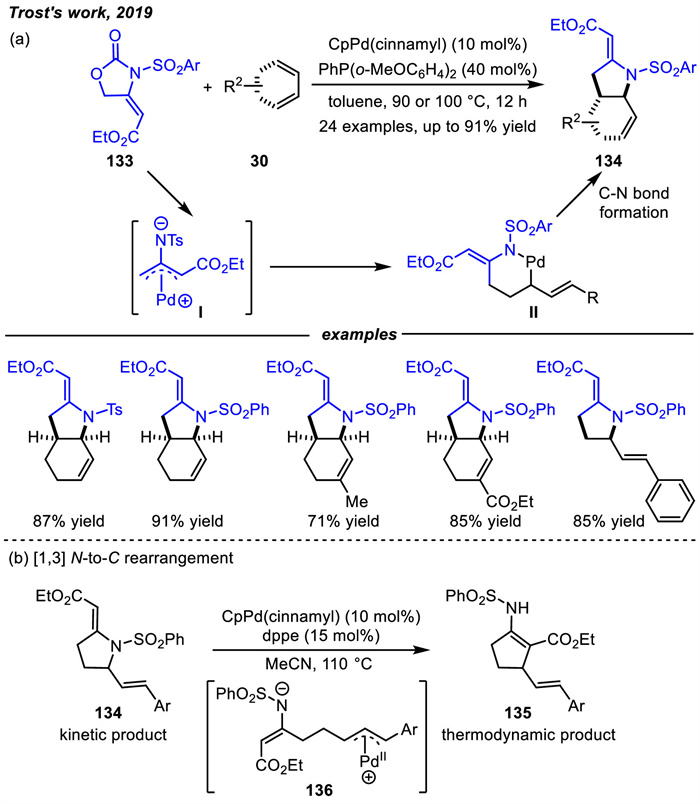
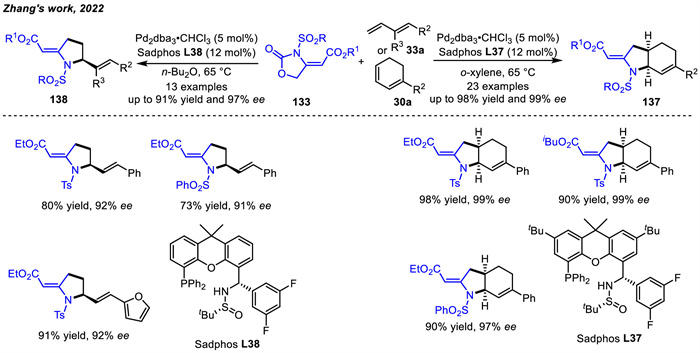
Additionally,Zhang and co-workers achieved highly substituted or fused pyrrolidines in asymmetric cycloaddition reactions of Pd-aminoallyl intermediates with 1,3-dienes in 2022 (Scheme 47) [75]. The cyclic or acyclic 1,3-dienes reactions with Pd-aminoallyl intermediates were catalyzed using chiral ligands Sadphos L37 or L38 respectively,in which aminoallyl precursors had a large range of ester groups,including Et,Me,nBu,iBu,Ph and Bn. Control experiments indicated that the apparent matched/mismatched effect of substrates was influenced by cis/trans-configuration of aminoallyl precursor and dienes. The cycloaddition products could not be obtained for E-133 and Z-33a. The asymmetric version required the aminoallyl precursor 133 with exclusive Z-geometry since it could reduce the activation entropy of the transition state generated by Pd/Sadphos-aminoallyl zwitterion. Moreover,a Pd-catalyzed [1,3] N-to-C rearrangement with the achiral ligand could also be achieved.
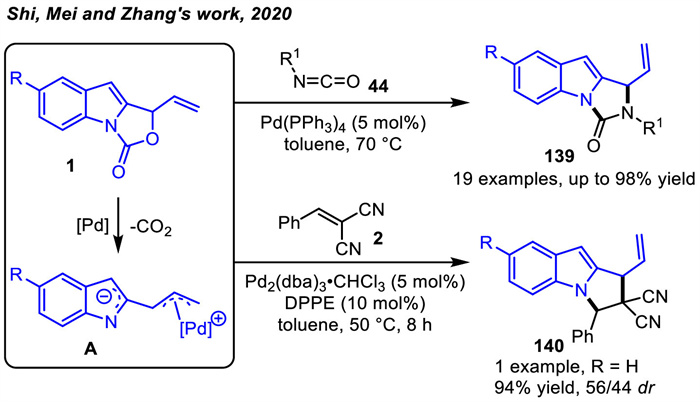
In 2020,Shi and coworkers [45] continued to expand the application of vinyl indoloxazolidones 1,which could be convert to Pd-π-allyl zwitterion intermediate A specifically used as a nitrogen-carbon-carbon building block (Scheme 48). Thus,palladium-catalyzed decarboxylative [3 + 2] cycloaddition reactions were achieved by reaction with isocyanates 44. Nevertheless,different chiral ligands (including chiral phosphoramidites and chiral bidentate phosphine ligands) could hardly control the enantioselectivity (< 8% ee) of the [3 + 2] cycloaddition reaction. Furthermore,when the electron-deficient olefin benzylidene malononitrile 2 was used as a dipolarophile for the reaction with oxazoloindol-3-one 1 under palladium catalysis,the product 140 was produced in 94% yield with a diastereoselectivity of 56/44.
Interestingly,in 2021,Chen and coworkers disclosed the first use of pyrrolidines 141 [132,133] for the palladium-catalyzed [5 + 2] cycloaddition reactions with azlactones or butenolides to produce seven-membered azepines 143 (Scheme 49) [134]. A wide range of substrate scope was suitable. R3 on 142 could be aryl,vinyl,alkyl. Moreover,the substituent on the nitrogen atom of substrates 141 could contain PMB,cyanide,phthalimide,or ester groups. A possible reaction pathway was depicted. Firstly,with the assistance of B(OH)3,the palladium catalyst underwent oxidative addition with 3-alkylidenepyrrolidines 141 to yield N-zwitterionic intermediate Ⅰ through an inert C-N bond cleavage. The substrates 142 acted as a two-atom synthon to capture intermediate Ⅰ to give intermediate Ⅱ. Subsequently,1,1-disubstituted alkene on intermediate Ⅱ may isomerized to form conjugated azlactone intermediate Ⅲ. The final azepines 143 were generated by an intramolecular lactamization step.

As described in this review,the palladium-catalyzed intermolecular cycloaddition reactions of 1,n-dipoles with different dipolarophiles in the present of ligands can be an effective and practical strategy for constructing structurally diverse 5- to 12-membered cyclic compounds,which are widely found in many natural products and pharmaceutical molecules and have received significant attention from synthetic chemists. We mainly summarize the progress of Pd-π-allyl zwitterions in the past 5 years,including newly designed Pd-π-allyl zwitterionics and several zwitterionics that have received renewed attention,which have also been designed and applied to diverse reaction partners to achieve new cyclic compounds that may be difficult to construct by other conventional methods.
Despite these great achievements allow for the rapid and convenient construction of complex polycyclics or medium-sized cycles exclusively with relatively simple substrates under mild conditions. There are several areas still need improvement. First of all,such transformations are often plagued by competitive reaction pathways and low levels of site- and stereoselectivity,and improvements in new catalytic systems may depend heavily on the design and discovery of new dipoles and chiral ligands. Secondly,the corresponding precursors of these key species are far from adequate,especially for 1,6- or 1,8-zwitterionic π-allyl palladium species,and their applications are still rare. Moreover,studies on the direct application of cycloaddition reactions via Pd-π-allyl zwitterion intermediates to the total synthesis of natural products,bioactive molecules,and pharmaceuticals are still scarce [135,136],even though this aspect is of great importance. Therefore,the design and discovery of novel zwitterionic π-allyl palladium species will be crucial to the continuous advancement of this field and the direct application of this reaction strategy to the synthesis of drug-containing or active molecules.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge the financial support from the National Natural Science Foundation of China (Nos. 22071143 and 21772215),Start-up Funding for Scientific Research of Nanning Normal University (No. 86612345) and "BAGUI Scholar" Program of Guangxi Province of China.
Z.X. Yu,Y. Wang,Y. Wang,Chem. Asian J. 5 (2010) 1072-1088. doi: 10.1002/asia.200900712
A. Otero,M.J. Chapela,M. Atanassova,J.M. Vieites,A.G. Cabado,Chem. Res. Toxicol. 24 (2011) 1817-1829. doi: 10.1021/tx200182m
W. Zhao,Z. Wang,J. Sun,Angew. Chem. Int. Ed. 51 (2012) 6209-6213. doi: 10.1002/anie.201200513
T. Nemoto,S. Harada,M. Nakajima,Asian J. Org. Chem. 7 (2018) 1730-1742. doi: 10.1002/ajoc.201800336
A. Lawer,J.A. Rossi-Ashton,T.C. Stephens,et al.,Angew. Chem. Int. Ed. 58 (2019) 13942-13947. doi: 10.1002/anie.201907206
R.L. Reyes,T. Iwai,M. Sawamura,Chem. Rev. 121 (2021) 8926-8947. doi: 10.1021/acs.chemrev.0c00793
X. Zhang,L. Lin,J. Li,et al.,Chin. J. Org. Chem. 41 (2021) 1878-1887. doi: 10.6023/cjoc202010026
J. Ling,M. Laugeois,V. Michelet,V. Ratovelomanana-Vidal,M.R. Vitale,Synlett 29 (2018) 928-932. doi: 10.1055/s-0036-1591540
R. Huisgen,Angew. Chem. Int. Ed. 2 (1963) 565-598. doi: 10.1002/anie.196305651
W. Sun,F. Jiang,H. Liu,et al.,Chin. Chem. Lett. 30 (2019) 363-366. doi: 10.3390/molecules24020363
H. Mehrabi,M. Hatami-Pour,Chin. Chem. Lett. 25 (2014) 1495-1498. doi: 10.1016/j.cclet.2014.05.024
G.Z. Yue,B. Liu,Chin. J. Org. Chem. 40 (2020) 3132-3153. doi: 10.6023/cjoc202005092
Z. Zhao,Z. Ou,S.J. Kalita,et al.,Chin. Chem. Lett. 33 (2022) 3012-3016. doi: 10.1016/j.cclet.2021.12.006
C.J. Easton,C.M.M. Hughes,G.P. Savage,G.W. Simpson,Cycloaddition reactions of nitrile oxides with alkenes,in: A.R. Katritzky (Eds. ),Advances in Heterocyclic Chemistry,Academic Press,New York,1994,pp. 261-327.
Y. Zhang,J. Shang,H. Li,et al.,Chin. Chem. Lett. 31 (2020) 1276-1280. doi: 10.1016/j.cclet.2019.10.039
B.M. Trost,D.M.T. Chan,J. Am. Chem. Soc. 101 (1979) 6429-6432. doi: 10.1021/ja00515a046
R. Shintani,M. Murakami,T. Hayash,J. Am. Chem. Soc. 129 (2007) 12356-12357. doi: 10.1021/ja073997f
N. De,E.J. Yoo,ACS Catal. 8 (2018) 48-58. doi: 10.1021/acscatal.7b03346
I. Shimizu,Y. Ohashi,J. Tsuji,Tetrahedron Lett. 26 (1985) 3825-3828. doi: 10.1016/S0040-4039(00)89261-8
B.D.W. Allen,C.P. Lakeland,J.P.A. Harrity,Chem. Eur. J. 23 (2017) 13830-13857. doi: 10.1002/chem.201702486
M. Meazza,H. Guo,R. Rios,Org. Biomol. Chem. 15 (2017) 2479-2490. doi: 10.1039/C6OB02647H
J. Wang,S.A. Blaszczyk,X. Li,W. Tang,Chem. Rev. 121 (2021) 110-139. doi: 10.1021/acs.chemrev.0c00160
B.M. Trost,P.J. Morris,S.J. Sprague,J. Am. Chem. Soc. 134 (2012) 17823-17831. doi: 10.1021/ja309003x
K.S. Halskov,L. Naesborg,F. Tur,K.A. Jorgensen,Org. Lett. 18 (2016) 2220-2223. doi: 10.1021/acs.orglett.6b00852
C.F. Xu,B.H. Zheng,J.J. Suo,C.H. Ding,X.L. Hou,Angew. Chem. Int. Ed. 54 (2015) 1604-1607. doi: 10.1002/anie.201409467
K. Spielmann,A. van der Lee,R.M. de Figueiredo,J. M. Campagne,Org. Lett. 20 (2018) 1444-1447. doi: 10.1021/acs.orglett.8b00228
S. Xiao,B. Chen,Q. Jiang,et al.,Org. Chem. Front. 8 (2021) 3729-3733. doi: 10.1039/d1qo00408e
C. Ma,Y. Huang,Y. Zhao,ACS Catal. 6 (2016) 6408-6412. doi: 10.1021/acscatal.6b01845
W.Y. Wang,J.Y. Wu,Q.R. Liu,et al.,Org. Lett. 20 (2018) 4773-4776. doi: 10.1021/acs.orglett.8b01886
J. Wang,L. Zhao,C. Zhu,et al.,Chin. Chem. Lett. 33 (2022) 4549-4558. doi: 10.1016/j.cclet.2022.01.063
C. Wang,J.A. Tunge,J. Am. Chem. Soc. 130 (2008) 8118-8119. doi: 10.1021/ja801742h
C. Guo,M. Fleige,D. Janssen-Muller,C.G. Daniliuc,F. Glorius,J. Am. Chem. Soc. 138 (2016) 7840-7843. doi: 10.1021/jacs.6b04364
L.A. Leth,F. Glaus,M. Meazza,et al.,Angew. Chem. Int. Ed. 55 (2016) 15272-15276. doi: 10.1002/anie.201607788
C. Guo,D. Janssen-Muller,M. Fleige,et al.,J. Am. Chem. Soc. 139 (2017) 4443-4451. doi: 10.1021/jacs.7b00462
M.M. Li,Y. Wei,J. Liu,et al.,J. Am. Chem. Soc. 139 (2017) 14707-14713. doi: 10.1021/jacs.7b08310
D. Mun,E. Kim,S.G. Kim,Synthesis 51 (2019) 2359-2370. doi: 10.1055/s-0037-1610685
S.N. Fairuz Binte Sheikh Ismail,B. Yang,Y. Zhao,Org. Lett. 23 (2021) 2884-2889. doi: 10.1021/acs.orglett.1c00505
C.H. Lu,S. Darvishi,V. Khakyzadeh,C. Li,Chin. Chem. Lett. 32 (2021) 405-407. doi: 10.1016/j.cclet.2020.02.056
J.J. Suo,J. Du,Y.J. Jiang,et al.,Chin. Chem. Lett. 30 (2019) 1512-1514. doi: 10.1016/j.cclet.2019.04.028
T.R. Li,Y.N. Wang,W.J. Xiao,L.Q. Lu,Tetrahedron Lett. 59 (2018) 1521-1530. doi: 10.1016/j.tetlet.2018.02.081
B. Niu,Y. Wei,M. Shi,Org. Chem. Front. 8 (2021) 3475-3501. doi: 10.1039/d1qo00273b
C. Yang,Z.X. Yang,C.H. Ding,B. Xu,X.L. Hou,Chem. Rec. 21 (2021) 1442-1454. doi: 10.1002/tcr.202000177
M.M. Zhang,B.L. Qu,B. Shi,W.J. Xiao,L.Q. Lu,Chem. Soc. Rev. 51 (2022) 4146-4174. doi: 10.1039/d1cs00897h
Y. You,Q. Li,Y.P. Zhang,et al.,ChemCatChem 14 (2022) e202101887. doi: 10.1002/cctc.202101887
Q.Q. Hang,S.J. Liu,L. Yu,et al.,Chin. J. Chem. 38 (2020) 1612-1618. doi: 10.1002/cjoc.202000104
F. Tian,W.L. Yang,T. Ni,J. Zhang,W.P. Deng,Sci. China Chem. 64 (2021) 34-40. doi: 10.1007/s11426-020-9854-3
Z. Zhao,X.X. Yang,G.Y. Ran,et al.,Org. Lett. 23 (2021) 4791-4795. doi: 10.1021/acs.orglett.1c01507
Z.Q. Rong,L.C. Yang,S. Liu,et al.,J. Am. Chem. Soc. 139 (2017) 15304-15307. doi: 10.1021/jacs.7b09161
Y. Wu,C. Yuan,C. Wang,et al.,Org. Lett. 19 (2017) 6268-6271. doi: 10.1021/acs.orglett.7b02704
S. Singha,T. Patra,C.G. Daniliuc,F. Glorius,J. Am. Chem. Soc. 140 (2018) 3551-3554. doi: 10.1021/jacs.8b00868
C. Yuan,Y. Wu,D. Wang,et al.,Adv. Synth. Catal. 360 (2018) 652-658. doi: 10.1002/adsc.201701247
H.W. Zhao,W.Q. Ding,L.R. Wang,et al.,Eur. J. Org. Chem. 2020 (2020) 5557-5562. doi: 10.1002/ejoc.202000914
G. Yang,Y.M. Ke,Y. Zhao,Angew. Chem. Int. Ed. 60 (2021) 12775-12780. doi: 10.1002/anie.202102061
B.M. Trost,Y. Wang,C.J. Hung,Nat. Chem. 12 (2020) 294-301. doi: 10.1038/s41557-019-0412-9
R. Shintani,S. Park,F. Shirozu,M. Murakami,T. Hayashi,J. Am. Chem. Soc. 130 (2008) 16174–16175. doi: 10.1021/ja807662b
R. Shintani,M. Murakami,T. Tsuji,H. Tanno,T. Hayashi,Org. Lett. 11 (2009) 5642-5645. doi: 10.1021/ol902326s
R. Shintani,T. Ito,M. Nagamoto,H. Otomo,T. Hayashi,Chem. Commun. 48 (2012) 9936-9938. doi: 10.1039/c2cc35259a
L.C. Yang,Y.N. Wang,R. Liu,et al.,Nat. Chem. 12 (2020) 860-868. doi: 10.1038/s41557-020-0503-7
C. Gao,X. Wang,J. Liu,X. Li,ACS Catal. 11 (2021) 2684-2690. doi: 10.1021/acscatal.0c05515
B.M. Trost,Z. Jiao,Y. Liu,C. Min,C.I.J. Hung,J. Am. Chem. Soc. 142 (2020) 18628-18636. doi: 10.1021/jacs.0c08348
B.M. Trost,Z. Jiao,J. Am. Chem. Soc. 142 (2020) 21645-21650. doi: 10.1021/jacs.0c11535
K. Li,S. Yang,B. Zheng,et al.,Chem. Commun. 58 (2022) 6646–6649. doi: 10.1039/d2cc01134d
A. Li,Y. Gao,J.B. Lu,et al.,Chem. Sci. 13 (2022) 9265-9270. doi: 10.1039/d2sc02985e
B.M. Trost,S. Schneider,J. Am. Chem. Soc. 111 (1989) 4430-4433. doi: 10.1021/ja00194a043
K. Ohe,H. Matsuda,T. Ishihara,et al.,J. Org. Chem. 58 (1993) 1173-1177. doi: 10.1021/jo00057a033
I. Ikeda,A. Ohsuka,K. Tani,T. Hirao,H. Kurosawa,J. Org. Chem. 61 (1996) 4971-4974. doi: 10.1021/jo951425k
B.M. Trost,Z. Huang,G.M. Murhade,Science 362 (2018) 564–568. doi: 10.1126/science.aau4821
J.J. Suo,J. Du,Q.R. Liu,et al.,Org. Lett. 19 (2017) 6658-6661. doi: 10.1021/acs.orglett.7b03386
J. Du,Y.J. Jiang,J.J. Suo,et al.,Chem. Comm. 54 (2018) 13143-13146. doi: 10.1039/c8cc07996j
K. Liu,I. Khan,J. Cheng,Y.J. Hsueh,Y.J. Zhang,ACS Catal. 8 (2018) 11600-11604. doi: 10.1021/acscatal.8b03582
Q. Cheng,F. Zhang,Y. Cai,Y.L. Guo,S.L. You,Angew. Chem. Int. Ed. 57 (2018) 2134-2138. doi: 10.1002/anie.201711873
Y. Zou,S. Chen,K.N. Houk,J. Am. Chem. Soc. 141 (2019) 12382-12387. doi: 10.1021/jacs.9b05762
W. Chai,Q. Zhou,W. Ai,et al.,J. Am. Chem. Soc. 143 (2021) 3595-3603. doi: 10.1021/jacs.0c13412
A. Buzas,F. Gagosz,Org. Lett. 8 (2006) 515-518. doi: 10.1021/ol053100o
W. Zhang,P.C. Zhang,Y.L. Li,H.H. Wu,J. Zhang,J. Am. Chem. Soc. 144 (2022) 19627-19634. doi: 10.1021/jacs.2c09799
B. Yan,L. Zuo,X. Chang,et al.,Org. Lett. 23 (2021) 351-357. doi: 10.1021/acs.orglett.0c03856
Y. Zheng,T. Qin,W. Zi,J. Am. Chem. Soc. 143 (2021) 1038-1045. doi: 10.1021/jacs.0c11504
A. Fanourakis,P.J. Docherty,P. Chuentragool,R.J. Phipps,ACS Catal. 10 (2020) 10672-10714. doi: 10.1021/acscatal.0c02957
R.J. Phipps,Synlett 27 (2016) 1024-1026. doi: 10.1055/s-0035-1561933
Q. Zhao,C. Chen,J. Wen,X.Q. Dong,X. Zhang,Acc. Chem. Res. 53 (2020) 1905-1921. doi: 10.1021/acs.accounts.0c00347
T.X. Liu,C. Zhang,P. Zhang,et al.,Org. Chem. Front. 9 (2022) 5564-5570. doi: 10.1039/d2qo01116f
X. Pan,L. Yu,S. Wang,et al.,Org. Lett. 24 (2022) 2099-2103. doi: 10.1021/acs.orglett.2c00290
L. Zuo,Y. Yang,W. Guo,Org. Lett. 23 (2021) 2013-2018. doi: 10.1021/acs.orglett.1c00148
Y. Yang,L. Zuo,K. Wei,W. Guo,Org. Lett. 23 (2021) 3195-3200. doi: 10.1021/acs.orglett.1c00929
T. Liu,Y. Fang,L. Zuo,et al.,Org. Chem. Front. 8 (2021) 1902-1909. doi: 10.1039/d1qo00070e
L. Zuo,P. Ma,T. Liu,et al.,Org. Lett. 23 (2021) 7330-7335. doi: 10.1021/acs.orglett.1c02401
Y. Liu,Y. He,Y. Liu,K. Wei,W. Guo,Org. Lett. 24 (2022) 2567-2572. doi: 10.1021/acs.orglett.2c00808
C. Larksarp,H. Alper,J. Org. Chem. 64 (1999) 4152-4158. doi: 10.1021/jo990430b
R.C. Larock,S.K. Stolz-Dunn,Tetrahedron Lett. 29 (1988) 5069-5072. doi: 10.1016/S0040-4039(00)80681-4
B. Guo,J.T. Njardarson,Chem. Comm. 49 (2013) 10802-10804. doi: 10.1039/c3cc46660d
B. Guo,G. Schwarzwalder,J.T. Njardarson,Angew. Chem. Int. Ed. 51 (2012) 5675-5678. doi: 10.1002/anie.201201367
H. Xu,S. Khan,H. Li,X. Wu,Y.J. Zhang,Org. Lett. 21 (2019) 214-217. doi: 10.1021/acs.orglett.8b03665
T. Bando,S. Tanaka,K. Fugami,Z.I. Yoshida,Y. Tamaru,Bull. Chem. Soc. Jpn. 65 (1992) 97-110. doi: 10.1246/bcsj.65.97
J. Du,Y.D. Hua,Y.J. Jiang,et al.,Org. Lett. 22 (2020) 5375-5379. doi: 10.1021/acs.orglett.0c01638
J. Wang,Y.F. Li,J. Du,et al.,Org. Lett. 24 (2022) 1561-1565. doi: 10.1021/acs.orglett.2c00253
J. Zhang,Y. Chen,Q. Wang,et al.,Chin. J. Org. Chem. 42 (2022) 3051-3101. doi: 10.6023/cjoc202206028
Y.N. Wang,L.C. Yang,Z.Q. Rong,et al.,Angew. Chem. Int. Ed. 57 (2018) 1596-1600. doi: 10.1002/anie.201711648
H. Uno,T. Imai,K. Harada,N. Shibata,ACS Catal. 10 (2020) 1454-1459. doi: 10.1021/acscatal.9b05377
H. Uno,K. Kawai,M. Shiro,N. Shibata,ACS Catal. 10 (2020) 14117-14126. doi: 10.1021/acscatal.0c03927
H. Xie,Z. Yang,L. Tang,et al.,Chem. Commun. 58 (2022) 10560-10563. doi: 10.1039/d2cc03666e
K.R. Lee,S. Ahn,S.G. Lee,Org. Lett. 23 (2021) 3735-3740. doi: 10.1021/acs.orglett.1c01135
I. Shimizu,Y. Ohashi,J. Tsuji,Tetrahedron Lett. 25 (1984) 5183-5186. doi: 10.1016/S0040-4039(01)81558-6
R. Shintani,K. Moriya,T. Hayashi,Chem. Commun. 47 (2011) 3057-3059. doi: 10.1039/c0cc05308b
Q. Liu,T.X. Liu,Y. Ru,X. Zhu,G. Zhang,Chem. Commun. 55 (2019) 14498-14501. doi: 10.1039/c9cc07950e
B. Mao,H. Liu,Z. Yan,et al.,Angew. Chem. Int. Ed. 59 (2020) 11316-11320. doi: 10.1002/anie.202002765
M.M. Li,B.L. Qu,Y.Q. Xiao,W.J. Xiao,L.Q. Lu,Sci. Bull. 66 (2021) 1719-1722. doi: 10.1016/j.scib.2021.04.037
Z.C. Chen,Z. Chen,Z.H. Yang,et al.,Angew. Chem. Int. Ed. 58 (2019) 15021-15025. doi: 10.1002/anie.201907797
Z. Chen,Z.C. Chen,W. Du,Y.C. Chen,Org. Lett. 23 (2021) 8559-8564. doi: 10.1021/acs.orglett.1c03279
G.S. Mahadik,S.R. Hitchcock,Tetrahedron Asymmetry 21 (2010) 33-38. doi: 10.1016/j.tetasy.2009.12.011
Z. Flegelová,M. Pátek,J. Org. Chem. 61 (1996) 6735-6738. doi: 10.1021/jo960756+
R.D. Gao,Q.L. Xu,B. Zhang,et al.,Chem. Eur. J. 22 (2016) 11601-11604. doi: 10.1002/chem.201602691
Z. Yuan,R. Pan,H. Zhang,et al.,Adv. Synth. Catal. 359 (2017) 4244-4249. doi: 10.1002/adsc.201700978
X. Song,L. Xu,Q. Ni,Org. Biomol. Chem. 18 (2020) 6617-6621. doi: 10.1039/d0ob01434f
Q. Li,R. Pan,M. Wang,H. Yao,A. Lin,Org. Lett. 23 (2021) 2292-2297. doi: 10.1021/acs.orglett.1c00420
A. Scuiller,X. Liu,M. Cordier,J. Garrec,A. Archambeau,Synlett 32 (2021) 981-986. doi: 10.1055/s-0040-1706038
J. Xu,W. Shi,M. Liu,et al.,Adv. Synth. Catal. 364 (2022) 2060-2066. doi: 10.1002/adsc.202200160
P.H. Dou,S.P. Yuan,Y. Chen,et al.,J. Org. Chem. 87 (2022) 6025-6037. doi: 10.1021/acs.joc.2c00276
M. Meazza,M. Kamlar,L. Jasikova,et al.,Chem. Sci. 9 (2018) 6368-6373. doi: 10.1039/c8sc00913a
M. Franc,I. Císařová,J. Veselý,Adv. Synth. Catal. 363 (2021) 4349-4353. doi: 10.1002/adsc.202100571
Q.L. Zhang,Q. Xiong,M.M. Li,et al.,Angew. Chem. Int. Ed. 59 (2020) 14096-14100. doi: 10.1002/anie.202005313
H. Dai,S.S. Ge,B.J. Dai,et al.,Chin. J. Org. Chem. 35 (2015) 2610-2616. doi: 10.6023/cjoc201508015
M. Garg,M. Chauhan,P.K. Singh,J.M. Alex,R. Kumar,Eur. J. Med. Chem. 97 (2015) 444-461. doi: 10.1016/j.ejmech.2014.11.051
X.X. Liang,L. Zhang,F.L. Li,et al.,Future Med. Chem. 12 (2020) 223-242. doi: 10.4155/fmc-2019-0294
C. Wang,J.A. Tunge,Org. Lett. 8 (2006) 3211-3214. doi: 10.1021/ol0610744
B.D. Allen,M.J. Connolly,J.P. Harrity,Chem. Eur. J. 22 (2016) 13000-13003. doi: 10.1002/chem.201602586
M. Suzuki,S. Yoshida,K. Shiraga,T. Saegusa,Macromolecules 31 (1998) 1716-1719. doi: 10.1021/ma971222t
V. Garcia-Vazquez,L. Hoteite,C.P. Lakeland,D.W. Watson,J.P.A. Harrity,Org. Lett. 23 (2021) 2811-2815. doi: 10.1021/acs.orglett.1c00752
I. Khan,B.H. Shah,C. Zhao,F. Xu,Y.J. Zhang,Org. Lett. 21 (2019) 9452-9456. doi: 10.1021/acs.orglett.9b03662
F. Jiang,F.R. Yuan,L.W. Jin,G.J. Mei,F. Shi,ACS Catal. 8 (2018) 10234-10240. doi: 10.1021/acscatal.8b03410
B.M. Trost,Z. Huang,Angew. Chem. Int. Ed. 58 (2019) 6396-6399. doi: 10.1002/anie.201900693
T. Yoshinao,K. Masanari,T. Shuji,K. Sigeru,Y. Zen-ichi,Bull. Chem. Soc. Jpn. 67 (1994) 2838-2849. doi: 10.1246/bcsj.67.2838
M. Mimura,M. Hayashida,K. Nomiyama,et al.,Chem. Pharm. Bull. 41 (1993) 1971-1986. doi: 10.1248/cpb.41.1971
J. Zhou,H. Liu,Z. Li,C. Jin,W. Su,Tetrahedron Lett. 58 (2017) 3174-3177. doi: 10.1016/j.tetlet.2017.07.002
L. Wu,T. Wang,C. Gao,et al.,ACS Catal. 11 (2021) 1774-1779. doi: 10.1021/acscatal.1c00130
X. Liang,T.Y. Zhang,X.Y. Zeng,et al.,J. Am. Chem. Soc. 139 (2017) 3364-3367. doi: 10.1021/jacs.7b00854
M. Li,Y. Liu,H. Si,X. Zhou,Y.J. Zhang,Org. Lett. 24 (2022) 7812-7816. doi: 10.1021/acs.orglett.2c03159

Scheme 12 General reaction process of Pd-catalyzed cycloadditions via Pd-oxyallyl intermediates.

Scheme 18 Pd-catalyzed enantioselective [3 + 2] cycloadditions of VMCCs with aryl ketones or isocyanates.

Scheme 29 General Pd-catalyzed cycloaddition of alkylidenetrimethylene carbonates (ADTMCs).

Scheme 35 The formation of oxa-1,4-dipole from ADTMC and 3-hydroxy-2-methylenepropyl methyl carbonate.

Scheme 36 Pd-catalyzed [4 + 2] cycloadditions of 3-hydroxy-2-methylenepropyl methyl carbonate.

Scheme 37 Pd-catalyzed [4 + 2] and [4 + 4] cycloadditions of tert-butyl[2-(hydroxymethyl)allyl] carbonate.

Scheme 39 Pd-catalyzed ring contraction/formal cycloadditions with pyrazolone derivatives.

Scheme 40 Pd-catalyzed ring contraction/formal cycloadditions with thiazole derivatives.

Scheme 44 Pd-catalyzed [4 + 2] cycloadditions of carbamate 115 with α-fluoro β-ketoesters or α-SCF3-substituted ketones.

Scheme 47 Pd-catalyzed asymmetric [3 + 2] cycloadditions via Pd-aminoallyl intermediates.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:





































