Structure-directed expansion of biphenyl-pyridone derivatives as potent non-nucleoside reverse transcriptase inhibitors with significantly improved potency and safety
Citation:
Li-Min Zhao, Christophe Pannecouque, Erik De Clercq, Shuai Wang, Fen-Er Chen. Structure-directed expansion of biphenyl-pyridone derivatives as potent non-nucleoside reverse transcriptase inhibitors with significantly improved potency and safety[J]. Chinese Chemical Letters,
2023, 34(12): 108261.
doi:
10.1016/j.cclet.2023.108261
Structure-directed expansion of biphenyl-pyridone derivatives as potent non-nucleoside reverse transcriptase inhibitors with significantly improved potency and safety
English
Structure-directed expansion of biphenyl-pyridone derivatives as potent non-nucleoside reverse transcriptase inhibitors with significantly improved potency and safety
Engineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai 200433, China
b.
Shanghai Engineering Center of Industrial Asymmetric Catalysis for Chiral Drugs, Shanghai 200433, China
c.
Key Laboratory of Natural Resources of Changbai Mountain & Functional Molecules, Ministry of Education, Yanbian University College of Pharmacy, Yanbian University, Yanji 133002, China
d.
Rega Institute for Medical Research, KU Leuven, Herestraat 49, Leuven B-3000, Belgium
☆ Dedication to Prof. Lixin Dai on the Occasion of His Centenary Birthday. * Corresponding authors. E-mail addresses: rfchen@fudan.edu.cn (F.-E. Chen)
Received Date:
10 January 2023 Accepted Date:
21 February 2023 Revised Date:
16 February 2023 Available Online:
15 December 2023
Abstract:
Following our previous work on human immunodeficiency virus-1 (HIV-1) non-nucleoside reverse transcriptase inhibitors (NNRTIs), a series of novel biphenyl-pyridone derivatives were synthesized and evaluated for their anti-HIV-1 activity to expand their structure–activity relationship. Some of them exhibited low nanomolar activity toward wild-type HIV-1 and clinically relevant single/double mutant strains. The most active compound B1 was 231-fold more potent (EC50 = 17 nmol/L) than the lead compound 2 (EC50 = 3.93 µmol/L) against wild-type (WT) HIV-1. This compound was approximately 3.5-fold less cytotoxic (CC50 = 100.58 µmol/L) than compound 2 (CC50 = 28.24 µmol/L), presenting a higher selectivity index (SI) value of 5923. Compared with 2, the antiviral potency of B1 was significantly increased against five single mutant strains (L100I, K103N, E138K, Y181C and Y188L) and two double mutant strains (F227L+V106A and K103N+Y181C). Especially, K103N, Y181C and K103N+Y181C were more sensitive to B1 than both 2 and doravirine. Besides, the enzymatic inhibitory activity of B1 against wild-type HIV-1 reverse transcriptase was approximately 32-fold higher (IC50 = 100 nmol/L) than 2 (IC50 = 3.21 µmol/L). Molecular docking studies and dynamic simulations were conducted to explain their potent activity. Taken together, this research represents an important step toward the discovery of novel biphenyl-pyridone drug candidates for HIV therapy.
Acquired immunodeficiency syndrome (AIDS), caused by human immunodeficiency virus (HIV) infection, is currently one of the major pandemics [1,2]. In 2021, more than 38.4 million people living with HIV have been reported globally, along with about 650,000 deaths [3]. Targeting reverse transcriptase has proven to be an effective strategy for HIV treatment [4-8]. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), a key component of active antiretroviral therapy (ART), effectively inhibit reverse transcriptase by targeting its allosteric binding site, which was located approximately 10 Å from the catalytic site of DNA polymerase [9-12]. Although the six Food and Drug Administration (FDA)-approved NNRTIs, such as etravirine (ETR) and doravirine (DOR), have achieved significant clinical efficacy in the treatment of HIV, the rapid emergence of drug resistance remains a critical issue, leading to treatment failure [13,14]. Therefore, continued efforts to d3evelop novel NNRTIs with good drug resistance and low toxicity are still urgently needed.
Recently, the displacement of 5-chloro-3-cyanophenyl of DOR with a larger biphenyl ring was first proposed by us to strengthen π-π stacking and hydrophobic interactions with surrounding aromatic hydrophobic residues Y188, Y181, F227 and W229, affording a series of biphenyl-pyridone derivatives [15]. Notably, transfer of the methyl group from 4-position of the biphenyl ring to 2-position resulted in a 2-fold increase in potency against wild-type (WT) HIV-1 with the 50% effective concentration (EC50) value of 3.93 µmol/L and slightly reduced cytotoxicity [the 50% cytotoxic concentration (CC50) = 33.82 µmol/L], as shown in Fig. S1 (Supporting information). The main difference in the docking results was that the biphenyl ring of compound 1 formed three π-π stacking interactions with W299 and Y181, while the biphenyl fragment of compound 2 not only maintained the same interactions with W299 and Y181 but also interacted with Y188 and Y183 via a π-π stacking interaction, respectively. The transfer of the methyl group from the 4-position to the 2-position enlarged the dihedral angle of the biphenyl ring, and oriented the molecule more favorably, thereby contributing to increasing the binding affinity between the target molecule 2 and the protein.
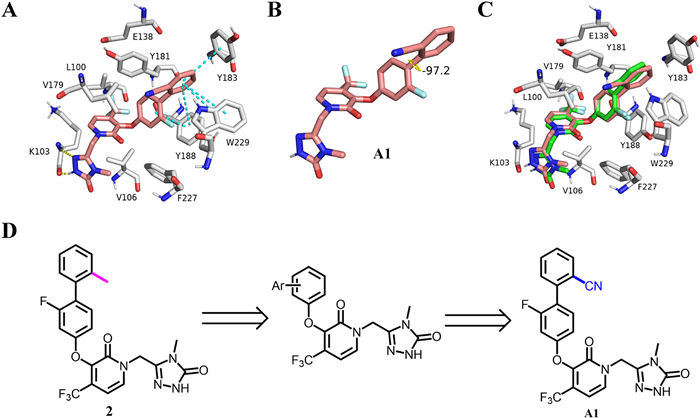
As part of our ongoing research on NNRTIs [16-21], we initiated this project to search for biphenyl-pyridone anti-HIV drug candidates with good drug-like properties. Ortho-substitution of different groups on the biphenyl ring was preserved in the following structural optimization, which may have potential conformational implications for enhanced potency. The molecular toxicity prediction using discovery studio suggested that the replacement of the ortho-methyl group of 2 with a CN group would boost its security (Table S1 in Supporting information). Molecular docking studies showed that the designed compound A1 was well projected into the binding pocket of WT HIV-1 reverse transcriptase (Fig. 1A). In addition to maintaining three π-π stacking interactions with W299 and Y183, compound A1 formed two π-π stacking interactions with Y188, which was different from 2, and its dihedral angle of the biphenyl ring was further enlarged (Fig. 1B). No other differences were observed in the superposition of A1 and 2 (Fig. 1C). Based on the above hypothesis, a series of novel biphenyl-pyridone derivatives were synthesized, and their anti-HIV-1 activity and cytotoxicity were evaluated.
Figure 1
Figure 1.
(A, B) Rendering of computed structure for A1 (pink) with wide type HIV-1 reverse transcriptase (PDB code: 4NCG); (C) Overlay of A1 and 2 within the binding pocket; (D) Structure-directed optimization for the parent biphenyl-pyridone 2.
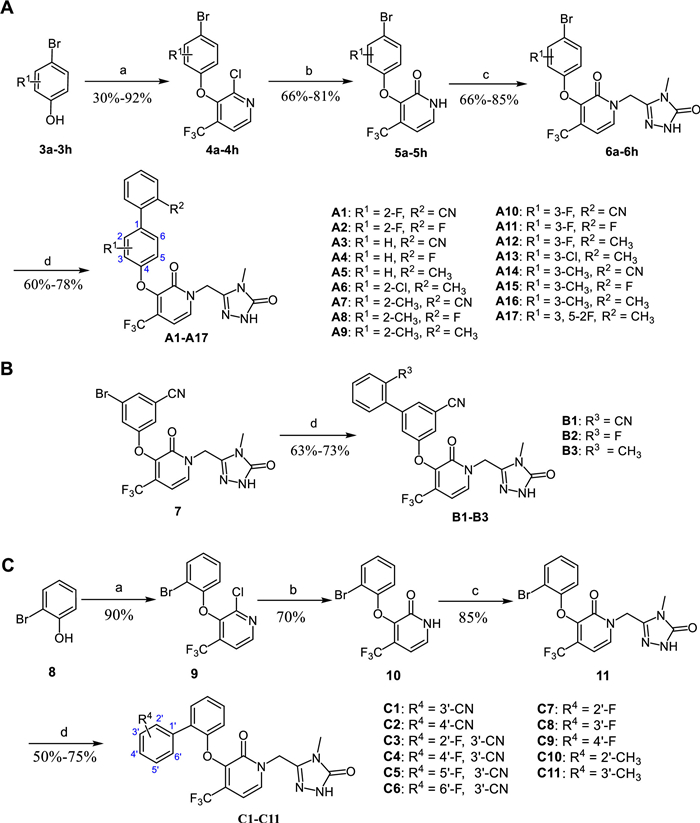
The synthetic route of the target compounds A1–A17 was depicted in Scheme 1A [15]. The nucleophilic substitution of the commercially available chloropyridine and appropriate p-bromophenols (3a–3h) was conducted using K2CO3 as base in NMP at 80 ℃ for 5 h, producing phenylchloropyridines 4a–4h in 30%–92% yields. Hydrolysis of the chloropyridine with NaOH resulted in hydroxypyridines 5a–5h in 66%–81% yields, followed by being treated with 5-(chloromethyl)-4-methyl-2,4-dihydro-3H-1,2,4-triazol-3-one in dry DMF to give key bromopyridine intermediates 6a–6h in 66%–85% yields. Finally, 6a–6h were then subjected to Suzuki coupling reaction, catalyzed by Pd(dppf)Cl2 at 110 ℃, delivering the desired products A1–A17 in 60%–78% yields [22]. Schemes 1B and C described the same synthetic approach of target compounds B1–B3 and C1–C11 as A1–A17.
All the newly synthesized compounds were evaluated for their antiviral activity and cytotoxicity in MT-4 cells infected with WT HIV-1 strain (IIIB), with DOR as the reference drug. The biological results were summarized in Tables 1 and 2 and expressed as EC50 (anti-HIV potency), CC50 (cytotoxicity), and selectivity index (SI, CC50/EC50 ratio).
Table 1
Table 1.
Activity and cytotoxicity of compounds A1–A17 against HIV-1 (IIIB) strains in MT-4 cells.
As shown in Table 1, the replacement of the ortho-methyl group of 2 with a CN group led to significant increase in potency toward WT HIV-1 (A1, EC50 = 0.36 µmol/L) and much lower cytotoxicity (CC50 = 121.58 µmol/L), which was consistent with our predicted results. Subsequently, the substitution of the ortho-methyl group with a fluorine substituent resulted in decreased activity (A2, EC50 = 7.77 µmol/L). When the 2-F substituent of A1 and A2 was removed, the activity of compounds A3 and A4 was significantly reduced, which was similar to the trend of compound 2 in deleting the 2-F substituent. Comparable activity was obtained by replacing the 2-F substituent of 2 with a chlorine substituent or a methyl group (A6 and A9). Introducing a methyl group to replace the fluorine substituent of A1 or A2 decreased their potency (A7 and A8). The incorporation of a fluorine substituent or a methyl group at the 3-position of A3–A5 did not improve their potency. Shifting the methyl group of A6 from the 2-position to the 3-position resulted in reduced activity (A13). Introducing a fluorine substituent to the 5-position of the biphenyl ring of A12 doubled its potency (A17, EC50 = 3.37 µmol/L), but did not alter cytotoxicity. Pleasingly, when the para-biphenyl group of A3–A5 was changed to the meta-biphenyl group, the potency of B1–B3 was significantly improved with nanomolar activity toward WT HIV-1 (IIIB) strain, as shown in Table 2. Especially, the most active compound B1 bearing an ortho-CN group possessed comparable activity to DOR with an EC50 value of 17 nmol/L, about 231-fold more potent than 2 (EC50 = 3.93 µmol/L). The cytotoxicity of B1 was three times lower (CC50 = 100.58 µmol/L) than 2 (CC50 = 28.24 µmol/L). In addition, compounds A1–A17 and B1–B3 displayed no inhibitory activity against HIV-2 strain (ROD) with EC50 values over 35 µmol/L.
Next, we further surveyed the effect of ortho-biphenyl group on activity and the results were collected in Table 2. Some of them, including C1, C2, C4 and C5, showed nanomolar inhibitory activity against WT HIV-1, but not as potently as B1. Moreover, most of them possessed unsatisfactory cytotoxicity and selectivity. The structure-activity relationship was as follows: C1 and C2 with a 3′-CN or 4′-CN group had similar inhibitory activity toward WT HIV-1, better than 2. The incorporation of fluorine atom at the 2′ and 6′ positions of C1 decreased its potency, but its activity slightly increased when fluorine atom was installed at the 4′ and 5′ positions of C1. Compared with 2, no obvious potency change was observed in compounds C7–C9 with a fluorine atom at the 2′, 3′, or 4′ position of the biphenyl ring. Replacing the fluorine atom of C7 and C8 with methyl group achieved no positive effect (C10 and C11).
The promising potency of compounds B1–B3 encouraged us to further evaluate their inhibitory activity against a panel of clinically observed single and double mutants (L100I, K103N, Y181C, Y188L, E138K, F227L+V106A, and K103N+Y181C), and the results were summarized in Table 3. B1–B3 had low nanomolar activity toward the tested HIV-1 mutant strains except for F227L+V106A. The most potent compound B1 possessed almost comparable inhibitory activity to DOR toward L100I, K103N, Y181C, Y188L, E138K, F227L+V106A, and K103N+Y181C. Notably, B1 was three- and six-fold more active than DOR against K103N and Y188L, respectively. A two-fold increase in the potency of B1 against K103N+Y181C was observed compared to DOR.
Table 3
Table 3.
Inhibitory activity of selected compounds toward clinically relevant HIV-1 mutant strains and WT HIV-1 RT.
The inhibitory activity of B1–B3 against WT HIV-1 reverse transcriptase was also assessed. The results were illustrated in Table 3. Compounds B1–B3 possessed nanomolar inhibitory activity toward WT HIV-1 reverse transcriptase, which was almost equivalent to DOR (IC50 = 44 nmol/L).
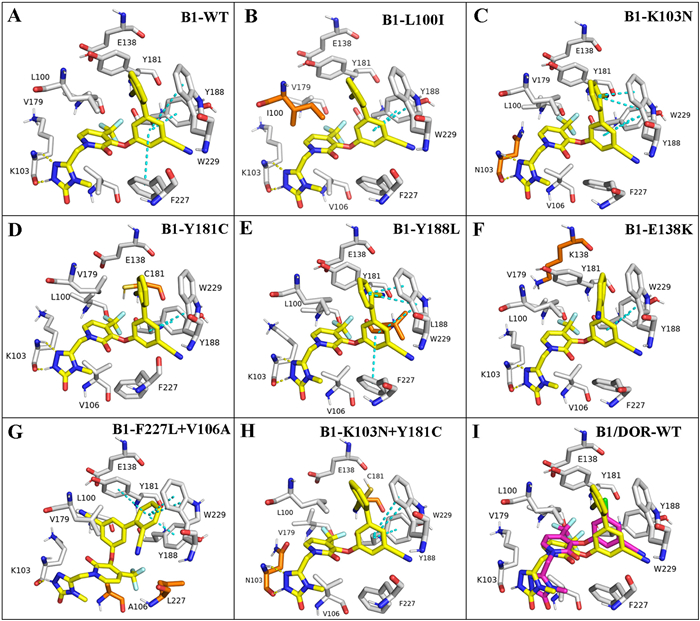
To characterize the binding mode of B1 within the non-nucleoside binding pocket of WT reverse transcriptase, molecular docking was carried out using the docking program Glide, and the protocol used was Induced Fit Docking. The structure of HIV-1 reverse transcriptase (RT) was downloaded from the PDB database (PDB code: 4NCG). The docking results were visualized by PyMOL. As shown in Figs. 2A and I, B1 was well positioned into the non-nucleoside reverse transcriptase inhibitors binding pocket and showed a similar binding conformation to that of DOR. The biphenyl fragment was inserted into the hydrophobic aromatic tunnel formed by Y181, Y188, F227 and W229, exhibiting positive π-π stacking interactions with W229, Y188 and F227, respectively. Two important hydrogen-bonding interactions between the triazolone moiety and amide K103 (C-N···H-N distance = 2.2 Å; H···O=C distance = 1.7 Å) were observed. Besides, B1 fitted well into the pockets of the L100I, K103N, Y181C, Y188L, E138K and K103N+Y181C mutants, respectively, and several common features (hydrogen bonds, and hydrophobic and electrostatic interactions) were still maintained as those in the WT RT enzyme (Figs. 2B–F and H). However, compound B1 adopted a fully flipped conformation in the binding pocket of F227L+V106A mutant strain (Fig. 2G), resulting in reduced potency due to the loss of the key hydrogen-bonding interactions with the K103 amide (C-N···H-N).
Figure 2
Figure 2.
Predicted binding modes of B1 (yellow) with HIV-1 RT and mutants (PDB code: 4NCG). (A) WT with B1; (B) L100I with B1; (C) K103N with B1; (D) Y181C with B1; (E) Y188L with B1; (F) E138K with B1; (G) F227L+V106A with B1; (H) K103N+Y181C with B1; and (I) WT with B1 (yellow) and DOR (pink). Mutated residues are depicted as orange sticks. Hydrogen bonds are depicted as yellow dashed lines and π-π bonds are shown as blue dashed lines.
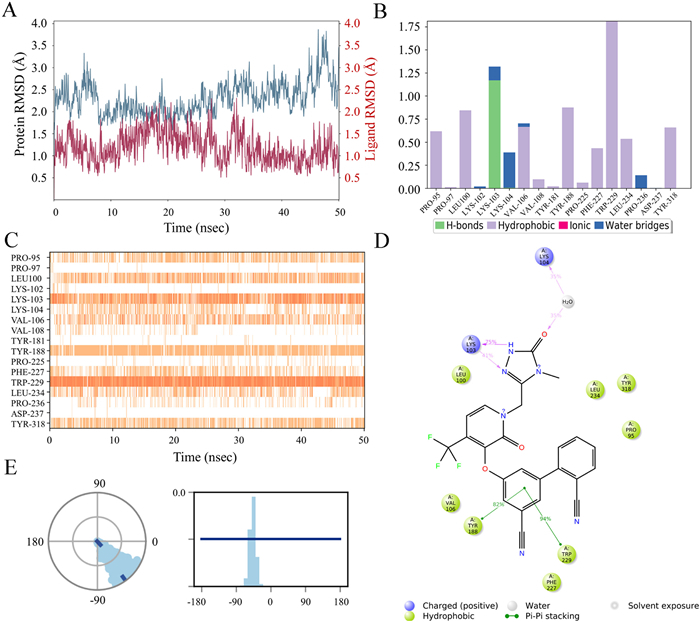
To gain further insight into the theoretical binding mode and docking complex stability of the best NNRTI inhibitors in this series, B1 was simulated for 50 ns using the software Schrödinger Maestro 11.4 (PDB code: 4NCG) with default settings to simulate its binding to WT HIV-1 reverse transcriptase. As shown in Fig. 3A, the root-mean-square deviation (RMSD) fluctuations of B1 were always smaller than that of the protein within 50 ns, indicating that the ligand B1 remained at its original binding site throughout the entire process. The 4NCG-B1 complex stabilized rapidly with the RMSD fluctuations below the allowable limit of 3 Å. Detailed schematic interactions of ligand B1 with the protein residues over 50 ns were presented in Figs. 3B–D. Four kinds of interactions, including hydrogen bonds, hydrophobic, ionic, and water bridges, were involved in the whole process of molecular dynamics simulation (Fig. 3B). 0.7 represents that the specific interaction spans 70% of 50 ns (Fig. 3B) and when multiple contacts between protein and ligand are possibly observed with values exceeding 1.0. A darker orange shade represents more than one specific contact of some residues with the ligand (Fig. 3C). Molecular dynamics simulations showed that B1 interacted with W229 viaπ-π stacking interactions for almost the entire 50 ns simulation time (Fig. 3C) and similar interactions between B1 and Y188 were also observed. The triazolone of B1 formed hydrogen bonds with K103 directly and a water-bridge hydrogen bond with K104. These interactions were almost consistent with the docking results in Fig. 3A. The ortho-substituted CN group expanded the dihedral angle of the biphenyl ring (Fig. 3E), which was favorable for maintaining a suitable conformation in the binding pocket.
Figure 3
Figure 3.
Molecular dynamics simulations analysis of compound B1 with WT HIV-1 reverse transcriptase (PDB code: 4NCG); (A) RMSD of compound B1; (B, C) Protein-ligand contacts analysis of MD trajectory; (D) Two-dimensional (2D) interaction diagram and interactions that occurred more than 30.0% of the simulation time in the selected trajectory; (E) The conformation of the torsion throughout the simulation (dial plot) and the probability density of the torsion (bar plot).
In summary, 31 new biphenyl-pyridone derivatives were synthesized and evaluated for their anti-HIV activity to enrich their structure-activity relationships. From this series prepared, compounds B1–B3 with different meta-substituted biphenyl groups had low nanomolar activity against wild-type HIV-1 and clinically observed single/double mutant strains. Of these, the most active compound B1 was comparable to doravirine (EC50 = 17 nmol/L) against WT HIV-1, which was 231-fold more potent than 2 (EC50= 3.93 µmol/L). Pleasingly, the cytotoxicity of B1 against MT-4 cells was significantly reduced (CC50 = 100 µmol/L) with a much higher SI value of 5923, compared with 2 (CC50 = 28.24 µmol/L, SI = 8.60). Besides, the compound B1 displayed nanomolar inhibitory activity against six clinically observed mutant strains (L100I, K103N, E138K, Y181C, Y188L and K103N+Y181C), and three of them, including K103N, Y181C and K103N+Y181C, were more sensitive to B1 than both 2 and doravirine. Furthermore, B1 possessed remarkably enhanced binding affinity to wild-type HIV-1 reverse transcriptase (IC50 = 100 nmol/L) compared to 2 (IC50 = 3.21 µmol/L). Molecular docking study revealed that introducing an ortho-substituted cyano group on the phenyl ring enlarged its dihedral angle, which facilitated the localization of biphenyl fragment to the aromatic-rich region in a more suitable manner and contributed to enhancing their biological activity viaπ-π stacking interactions with W229, Y188 and F227. Furthermore, two crucial hydrogen-bonding interactions of the triazolone moiety with residue K103 were retained. Also, molecular dynamic simulations demonstrated the stability of the protein-ligand complex throughout 50 ns. The discovery of B1 represented a promising starting point in the development of even more efficacious anti-HIV drugs.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (No. 22077018). We also thank Fudan University for providing the sources of molecular modeling.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108261.
[1]
E. De Clercq, Nat. Rev. Drug Discov. 6 (2007) 1001–1018. doi: 10.1038/nrd2424
[2]
A.S. Fauci, H.C. Lane, N. Engl. J. Med. 383 (2020) 1–4. doi: 10.1056/nejmp1916753
[3]
https://www.unaids.org/en (The latest data on HIV released by UNAIDS in 2022).
[4]
C. Beyrer, A. Pozniak, N. Engl. J. Med. 377 (2017) 1605–1607. doi: 10.1056/NEJMp1710608
Figure 1
(A, B) Rendering of computed structure for A1 (pink) with wide type HIV-1 reverse transcriptase (PDB code: 4NCG); (C) Overlay of A1 and 2 within the binding pocket; (D) Structure-directed optimization for the parent biphenyl-pyridone 2.
Figure 2
Predicted binding modes of B1 (yellow) with HIV-1 RT and mutants (PDB code: 4NCG). (A) WT with B1; (B) L100I with B1; (C) K103N with B1; (D) Y181C with B1; (E) Y188L with B1; (F) E138K with B1; (G) F227L+V106A with B1; (H) K103N+Y181C with B1; and (I) WT with B1 (yellow) and DOR (pink). Mutated residues are depicted as orange sticks. Hydrogen bonds are depicted as yellow dashed lines and π-π bonds are shown as blue dashed lines.
Figure 3
Molecular dynamics simulations analysis of compound B1 with WT HIV-1 reverse transcriptase (PDB code: 4NCG); (A) RMSD of compound B1; (B, C) Protein-ligand contacts analysis of MD trajectory; (D) Two-dimensional (2D) interaction diagram and interactions that occurred more than 30.0% of the simulation time in the selected trajectory; (E) The conformation of the torsion throughout the simulation (dial plot) and the probability density of the torsion (bar plot).

 DownLoad:
DownLoad:




 下载:
下载:





