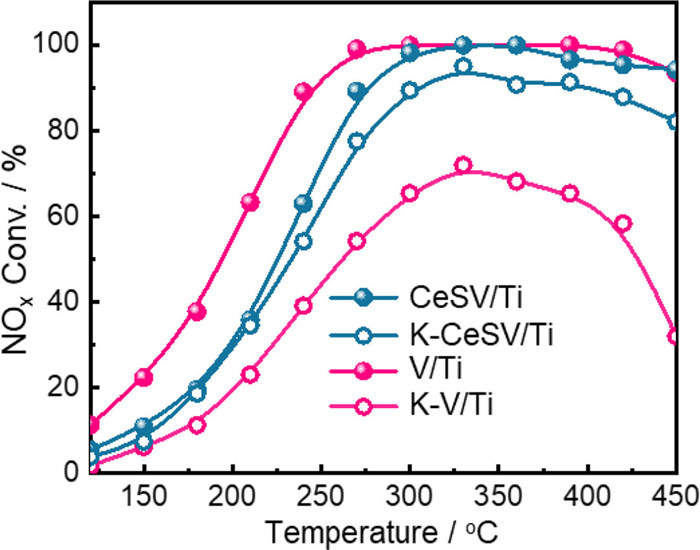
Figure 1.
Plots of NOx conversion versus temperature over CeSV/Ti, K-CeSV/Ti, V/Ti and K-V/Ti catalysts. Reaction conditions: 500 ppm NO, 500 ppm NH3, 5 vol% O2, N2 as the balance gas, and GHSV of 50,000 h−1.
NOx reduction against alkali poisoning over Ce(SO4)2-V2O5/TiO2 catalysts by constructing the Ce4+–SO42− pair sites
Shuangxi Li , Huijun Yu , Tianwei Lan , Liyi Shi , Danhong Cheng , Lupeng Han , Dengsong Zhang

NOx emissions from industries and vehicles not only impair human health but also cause serious environmental issues such as PM 2.5 and O3 [1-3]. NOx from power plants could be effectively reduced by NH3 selective catalytic reduction (NH3-SCR) using V2O5-WO3/TiO2 catalysts [4-6]. Although commercial V2O5-based catalysts have excellent activity and SO2 resistance at high temperatures (>300 ℃), the unsatisfactory alkali-resistant capacity restricts their broader applications in steel plants, biomass burning, waste incineration plants, etc. [7-10]. Therefore, a large number of studies have been conducted to explore effective strategies to ameliorate the alkali-resistance of catalysts.
In general, the deactivation mechanisms of alkali poisoning include the blockage of catalyst channels and the decrease in surface acidity, which diminishes the adsorption and activation of NH3. Extensive research has been studied on the improvement of alkali tolerance via increasing more acidic sites. Sulfated TiO2 and ZrO2 improved the acidity of catalysts and thus effectively improved alkali resistance [11-13]. The ion-exchanged titanate nanotubes (TNTs) have a number of OH groups with adequate acid content and could neutralize the basicity of alkali. Some TNTs-based catalysts such as CeO2-doped sulfated TNTs [14], Mo modified V-TNTs [15] and Nb-doped Ce nanotubes [16] showed excellent alkali-resistance. Furthermore, it is effective to enhance the alkali-resistance of catalysts via constructing alkali-trapping sites to protect active sites. It has been demonstrated that BO33− and PO43− as capturing sites could preferentially bond with alkali metals and protect active sites of CeO2 and Fe2O3 based catalysts [17,18]. Zhou et al. [9] found that the SCR activity was well maintained over the K-poisoned V2O5/CeO2 because the in situ constructed SO42– sites could capture K and retain a high adsorption rate and reactivity of NH3. Fe2(SO4)3/TiO2 catalysts are also highly resistant to alkali due to the protective effects of SO42– species [19]. The reactivity of K-poisoned catalysts could be recovered via SO2 treatment because SO2 preferred to combine with K, thus releasing the active sites poisoned by K [6]. These results demonstrated the protective effects of SO42– species against alkali-poisoning. Recently, we found that Ce4+–SO42− pair sites on S doped CeO2 catalysts not only increased the Brønsted acid sites but also greatly improved the SO2-resistance of catalysts resulting from suppressing the adsorption and oxidation of SO2. It is promising to develop alkali-resistant catalysts by constructing Ce4+–SO42− pair sites, which is of significance to the practice application of catalysts in complex working conditions.
In this study, we demonstrated that Ce(SO4)2 modified V2O5/TiO2 (denoted as CeSV/Ti) catalysts exhibited improved NOx reduction against alkali poisoning than V2O5/TiO2 (denoted as V/Ti). K poisoned V/Ti showed poor activity with NOx conversion below 70%. In comparison, K-poisoned CeSV/Ti showed much higher activity with NOx conversion above 80% within ~270–450 ℃. For revealing the promoting effects of Ce(SO4)2 on the K-resistant capacity of V/Ti catalysts, the structure features of catalysts are investigated by X-ray diffraction (XRD); the acidic amounts of catalysts were determined by NH3 temperature-programmed desorption combined with online mass spectrum (NH3-TPD-MS), and redox properties probed of H2 temperature-programmed reduction (H2-TPR), X-ray photoelectron spectroscopy (XPS), etc. In addition, the adsorption and activation of NH3 and NO as well as the pathways were deeply investigated by in situ diffuse reflectance infrared fourier transform spectra (DRIFTS).
The catalysts were prepared via an impregnation method. Take the preparation of 10 wt% Ce(SO4)2 modified V2O5/TiO2 catalyst as an example. First, 2 g of TiO2 was dispersed in 10 mL of deionized water and sonicated for 5 min. 0.2 g Ce(SO4)2 and 0.051 g NH4VO3 were dissolved in 6 mL of deionized by a heating method, respectively. After the mixed solution was cooled, the Ce(SO4)2 solution was added by drop to the TiO2 suspension and stirred for 1 h and then the NH4VO3 solution was added by drop. After stirring for 30 min, the samples are dehydrated using a rotary evaporator and then dried overnight in an oven at 80 ℃. After the dried sample was calcined in a muffle furnace at 450 ℃ for 4 h with a ramping rate of 2 ℃/min, 10 wt% Ce(SO4)2 modified V2O5/TiO2 was obtained, named as CeSV/Ti. Other 5, 15 and 20 wt% Ce(SO4)2 modified V2O5/TiO2 catalysts were prepared using the same method. The Ce4+-modified and SO42−-modified V/Ti catalysts were prepared by impregnating 0.2614 g of Ce(NO3)3·6H2O and 0.1591 g of (NH4)2SO4, respectively. The preparation of V2O5/TiO2 catalysts is similar to CeSV/Ti without the addition of Ce(SO4)2, named as V/Ti. K-poisoned CeSV/Ti and V/Ti catalysts were prepared by the impregnation method. 0.017 g of KNO3 was dissolved in 10 mL of deionized water, and 0.5 g CeSV/Ti and V/Ti catalysts were added and stirred for 1 h. The mixture was dehydrated by a rotary evaporator, dried overnight in an oven at 80 ℃, and then calcined in a muffle furnace under 450 ℃ for 4 h with a ramping rate of 2 ℃/min. The obtained catalysts were named as K-CeSV/Ti and K-V/Ti, respectively. Characterizations of the obtained catalysts can be found in Supporting information.
SCR activity was characterized by using a fixed-bed quartz flow reactor (inner diameter: 8 mm) with the catalyst (40–60 meshes). SCR activity was tested from 150 ℃ to 420 ℃. The gas mixture composition: 500 ppm of NO, 500 ppm of NH3, 5 vol% of O2, and N2 as the balance gas. The gas hourly space velocity (GHSV) was controlled at 50,000 h−1 corresponding to the total flow rate of 257.7 mL/min. FTIR spectrometer (Protea atmosFIR) was used to simultaneously monitor the concentrations of NO, NO2, NH3, H2O, and N2O. The SCR activity data were recorded after the reaction system reached a stable state.
The NOx conversion, N2 selectivity, and GHSV were calculated by using the following formulas:
|
|
where NOx represents the total concentration of NO and NO2. [NOx]in, [NOx]out, [N2O]out, [NH3]in, and [NH3]out stood for the inlet and outlet concentrations of corresponding gas, respectively.
The GHSV was calculated by the following equation: GHSV=qv/πhr2qv meant the total flow rate; h represented the height of the catalyst in the reactor; and r stood for the inner radius of the reactor.
Firstly, the SCR activities of fresh and K-poisoned V/Ti catalysts were tested, while different amounts of Ce(SO4)2 modified V/Ti catalysts were compared (Figs. S1 and S2 in Supporting information). It can be seen that V/Ti shows NOx conversion above 90% within 240–450 ℃ (Fig. 1 and Fig. S1). Different amounts of Ce(SO4)2 modified V/Ti catalysts all show decreased activities to some extent, in which 10–15 wt% Ce(SO4)2 modified ones show satisfactory activity with NOx conversion above 90% from 270 ℃ to 450 ℃. After K-poisoning, the activity of K-V/Ti decreases markedly with the highest NOx conversion of only 72% at 330 ℃ (Fig. 1 and Fig. S2). For K-poisoned Ce(SO4)2 modified V/Ti catalysts, the activity increases and then decreases with increasing Ce(SO4)2 amount from 5 wt% to 20 wt%, and the 10 wt% Ce(SO4)2 modified V/Ti exhibits the highest activity with NOx conversion above 90% from 300 ℃ to 450 ℃ (Fig. 1). Besides, the fresh and K-poisoned CeSV/Ti show good N2 selectivity above 95% (Fig. S3 in Supporting information). The CeSV/Ti catalyst exhibits excellent H2O tolerance at 300 ℃ (Fig. S4 in Supporting information). Clearly, Ce(SO4)2 modification notably improves NOx reduction against alkali poisoning over V/Ti catalysts. The Ce(SO4)2 amount more than 10 wt% may led to the poor dispersion of Ce(SO4)2, that is unfavorable for the alkali resistance. Besides, we also supplemented the SCR activity for Ce4+-modified and SO42− modified V/Ti catalysts and their K-poisoned ones (Fig. S3). Although the low-temperature activities of CeV/Ti and SV/Ti are better than that of CeSV/Ti, the activity of K-poisoned CeSV/Ti is higher than that of K-poisoned CeV/Ti and SV/Ti catalysts. These results indicate that Ce4+–SO42− pair sites play crucial roles in the strong alkali resistance of CeSV/Ti.
Firstly, the thermal stability of CeSV/Ti catalyst was tested via the N2 temperature-programmed decomposition (TPDC (Fig. S5 in Supporting information). A small quantity of SO2 (m/z = 64) was detected below 500 ℃ that is derived from the decomposition of surface adsorbed SO42− species. The decomposition peak of Ce(SO4)2 occurs above 500 ℃ associated with the large formation of SO2. The thermogravimetry (TG) analysis also shows the similar results (Fig. S6 in Supporting information). These results indicate the good thermal stability of CeSV/Ti within the temperature range of the activity test of catalysts. In order to probe the nature reason for the stronger alkali-resistance of CeSV/Ti catalysts, the structure features of fresh and K-poisoned CeSV/Ti and V/Ti catalysts were investigated by XRD (Fig. S7 in Supporting information). All catalysts display characteristic diffraction peaks ascribed to anatase TiO2 (PDF #21–1272). There are no characteristic diffraction peaks related to Ce and V species, implying the highly dispersed state of Ce and V species. From the N2 adsorption-desorption isotherm, all fresh and K-poisoned catalysts show mesoporous features due to the unique capillary adsorption phenomenon (Figs. S8–S11 in Supporting information). It can be seen that the pore volumes and pore sizes of both K-poisoned catalysts decrease compared to the fresh ones (Table S1 in Supporting information), indicating that the K2O nanoparticles enter the pore channels and occupy partial space of pores of fresh catalysts. However, the specific surface areas of CeSV/Ti and V/Ti catalysts both increase to some extent after K-poisoning, which is likely owing to that K2O nanoparticles in the pore channels contribute to the specific surface areas. Besides, the pore volume and pore diameter of catalysts both decrease to some extent after K-poisoning, which results from that K2O nanoparticles occupy the partial position of pores surface. These results indicate that there are no correlations between textual features and the activity of catalysts.
Acid sites of catalysts play crucial roles in NH3 adsorption/activation, and NH3-TPD-MS was performed to study the acidic properties of fresh and K-poisoned catalysts. As shown in Fig. 2a, both CeSV/Ti and V/Ti catalysts show two NH3 desorption peaks, where the low-temperature peaks are attributed to the weak acid sites and the high-temperature one ascribed to strong acid sites, respectively [20,21]. It is notable that the total acid amount of V/Ti increases from 20.2 µmol/g to 26.0 µmol/g after Ce(SO4)2 modification. After K-poisoning, the total acid amount of K-CeSV/Ti and K-V/Ti both decreases to some extents. The total acid amount (12.0 µmol/g) of K-CeSV/Ti is higher than that (7.4 µmol/g) of K-V/Ti (Table S2 in Supporting information), which contributes to the SCR activity of K-CeSV/Ti. The reducibility of fresh and K-poisoned CeSV/Ti and V/Ti catalysts were probed by H2-TPR. As shown in Fig. 2b, the reduction peak at 447 ℃ on V/Ti is attributed to the reduction of VOx species. For CeSV/Ti, the reduction peak at 489 ℃ is ascribed to the reduction of VOx and Ce4+ species while the peaks at 546 and 640 ℃ are related to the reduction of SO42− species [22]. Compared with V/Ti, the higher reduction temperature of V species over CeSV/Ti indicates the strong interaction between Ce and V. After K-poisoning, the reduction peak of VOx species shifts to a higher temperature of 503 ℃ for K-V/Ti, indicating the K bonded VOx species become more difficult to reduce. The reduction temperatures of VOx, Ce4+ and SO42− species over K-CeSV/Ti have no large changes compared with CeSV/Ti. However, it is notable that the reduction peak intensity of Ce4+ and SO42− species markedly reduces over the K-poisoned CeSV/Ti, implying that SO42− species as sacrificial sites bond with K.
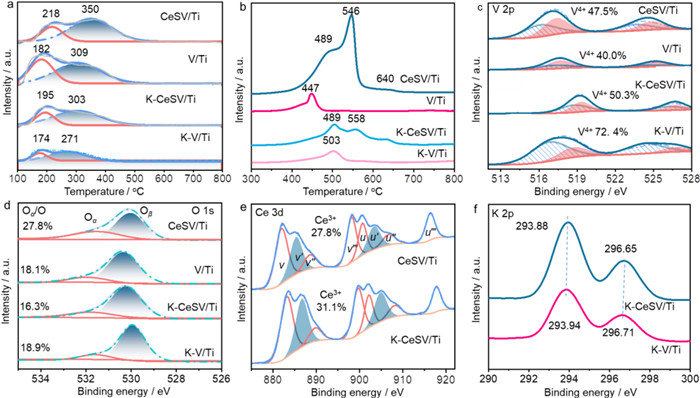
To investigate the redox properties of fresh and K-poisoned catalysts, XPS was applied to probe the electron states of V and O on the surface of catalysts. As shown in Fig. 2c, the XPS spectra of V 2p show two characteristic binding energies of V 2p3/2 and V 2p1/2, where the peaks around 516 and 524 eV are attributed to V4+ species while the peaks around 517 and 525 eV are ascribed to V5+ [23]. It can be found that the V4+/(V4++V5+) ratio (47.5%) on CeSV/Ti is higher than that (40.0%) on V/Ti, evidencing the strong interaction between Ce and V species over CeSV/Ti. The lower valence of V over CeSV/Ti indicates that the electron could transfer from Ce to V. After K-poisoning, the V4+/(V4++V5+) of K-V/Ti increases markedly to 72.4% while that (50.3%) of K-CeSV/Ti hardly changes compared to the fresh catalysts. The significant increase in V4+ fraction of K-V/Ti is attributed to the electron-donating effects of K that increase the electron density of V species. This also demonstrates that K strongly interacts with V species after K-poisoning. Differently, the maintenance of V valence over K-CeSV/Ti implies that K strongly interacts with Ce(SO4)2 rather than V. The O 1s XPS spectra were used to investigate the varieties and contents of surface oxygen species. The surface oxygen atomic percent (53.8%) of CeSV/Ti is much higher than that (27.0%) of V/Ti, which is attributed to the oxygen species of Ce(SO4)2 (Table S3 in Supporting information). After K-poisoning, the surface oxygen atomic percent of K-CeSV/Ti slightly decreases to 46.5% while that of K-V/Ti increases to 45.6%. These results imply that K2O is mainly bonded on Ce(SO4)2 sites of K-CeSV/Ti but cover on the surface of K-V/Ti. As shown in Fig. 2d, there are adsorbed oxygen (Oα) species around 531 eV and lattice oxygen (Oβ) species around 530 eV [24-31]. Ce(SO4)2 modification increases the Oα fraction of V/Ti from 18.1% to 27.8%, likely due to the contribution of surface SO42−. After K-poisoning, the Oα species of K-CeSV/Ti decrease notably to 16.3% while that of K-V/Ti keeps unchanged at 18.9%, respectively. It can be inferred that SO42− of Ce(SO4)2 strongly interacts with K over K-CeSV/Ti, which leads to the decrease of Oα species. By the contrast, K2O covers on the surface of K-V/Ti that results in the unchanged amount of Oα species because of the Oα species of K2O. In order to probe the interaction between Ce and K species on the K-CeSV/Ti catalysts, the XPS spectra of Ce 3d and K 2p are investigated over CeSV/Ti, K-CeSV/Ti and K-V/Ti catalysts (Figs. 2e and f). The peaks labelled as v, v'', v''' and u, u'' and u''' at 882, 888.9, 898, 901, 907 and 916.8 eV are attributed to the 3d104f0 Ce4+ state, while the peaks labeled v′ and u′ observed at 885 and 903 eV are ascribed to the 3d104f1 Ce3+ state (Fig. 2e) [28]. It can be found that the ratio of Ce3+/(Ce3++Ce4+) (31.1%) of K-CeSV/Ti is higher than that (27.8%) of CeSV/Ti, indicating the more electron-rich state of Ce in K-CeSV/Ti. On the other hand, the XPS spectra of K 2p were also investigated over K-CeSV/Ti and K-V/Ti catalysts. As seen in Fig. 2f, the binding energy of K on K-CeSV/Ti is higher than that on K-V/Ti, indicating the electron deficient state of K on the former one. Based on these results, it can be inferred that K is bonded on Ce(SO4)2 sites, leading to the transfer of electron from K to Ce species.
To explore the effects of K-poisoning on the adsorption and activation behavior of NH3 and NOx, the in situ DRIFTS of adsorption and desorption for NOx and NH3 species were investigated over fresh and poisoned catalysts. As shown in Fig. 3a, after introducing NH3, the band at 1429 cm−1 [32] ascribed to NH4+ and the bands around 1324 [33] and 1257 cm−1 [34] ascribed to NH3 species are observed on CeSV/Ti catalysts. These NH3/NH4+ species gradually decrease and disappear above 300 ℃ with increasing the desorption temperature. Similarly, the NH3 species (1328 and 1229 cm−1) [33,35] and NH4+ species (1470 and 1411 cm−1) [36,37] appear on V/Ti after the adsorption of NH3 (Fig. 3b). With the increasing desorption temperature, the NH3/NH4+ species gradually decrease and disappear above 300 ℃. Notably, there are more NH4+ species on CeSV/Ti catalysts and a larger number of NH3 species on V/Ti catalysts. After K-poisoning, the adsorption amount of NH3/NH4+ species both decrease on K-CeSV/Ti and K-V/Ti catalysts compared with the fresh catalysts. As seen in Fig. 3c, the NH3 species (1228 and 1321 cm−1) [33,35] and NH4+ species (1423 and 1467 cm−1) [38,39] appear on K-CeSV/Ti after the adsorption of NH3. These NH3/NH4+ species decrease and disappear above 260 ℃ with increasing the desorption temperature. As for K-V/Ti (Fig. 3d), NH3 species (1217 cm−1) [40] and NH4+ species (1398 and 1468 cm−1) [36,41] appear after the adsorption of NH3. With increasing desorption temperature, these NH3/NH4+ species decrease and disappear above 230 ℃. These results imply that the adsorption strength of NH4+ species on K-CeSV/Ti is stronger than that of NH3 species on K-V/Ti.
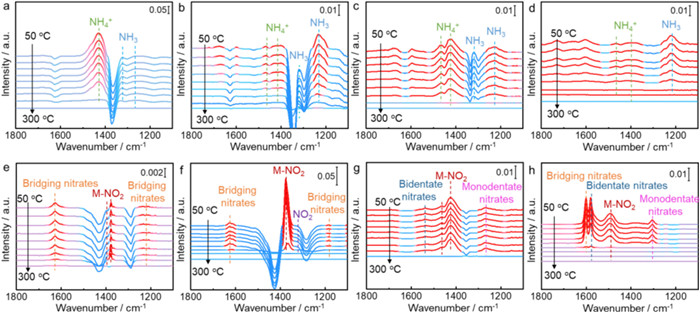
As shown in Fig. 3e, the adsorbed NOx species such as the bridging nitrate species (1626 and 1225 cm−1) and M-NO2 (1379 cm−1) appear on CeSV/Ti after the adsorption of NO+O2 [42]. With increasing desorption temperature, these NOx species gradually decrease and disappear above 300 ℃. As for V/Ti (Fig. 4f), the bridging nitrate species (1624 and 1183 cm−1), M-NO2 (1375 cm−1) and gaseous NO2 species (1324 cm−1) appear after the adsorption of NO+O2 [42]. These NOx species decrease and disappear above 260 ℃ with increasing the desorption temperature. It is notable that the amount of NOx species over V/Ti is much higher than that over CeSV/Ti. Besides, the NOx species adsorbed much more strongly on CeSV/Ti than that on V/Ti, implying that these NOx species on CeSV/Ti are not easy to participate in the SCR reaction. Therefore, the SCR reaction may occur on the CeSV/Ti catalyst between the adsorbed NHx species and gaseous NO following the Eley-Rideal (E-R) mechanism while the reaction might procced between the adsorbed NHx and NOx species following the Langmuir-Hinshelwood (L-H) mechanism over V/Ti. After K-poisoning, the adsorption strength of NOx species changes compared to fresh catalysts. As shown in Fig. 4g, the bidentate nitrate species (1543 cm−1), M-NO2 (1423 and 1466 cm−1) and monodentate nitrates (1264 cm−1) appear on K-CeSV/Ti after the adsorption of NO+O2 [42]. The amount of these NOx species is much more than that on fresh CeSV/Ti while these NOx species adsorb strongly and only disappear above 300 ℃ with increasing the desorption temperature. This result implies that K-poisoning improves the adsorption of NOx species but these species are relatively inactive. The reaction might still follow between NH4+ species and gaseous NO via the E-R mechanism over K-CeSV/Ti. As for K-V/Ti (Fig. 4h), the bridging nitrate species (1601 cm−1), bidentate nitrate species (1582 cm−1), M-NO2 (1493 cm−1) and monodentate nitrates (1307 cm−1) appear after the adsorption of NO+O2 [42]. The amount of these NOx species is much less than fresh V/Ti. With the increase of desorption temperature, these NOx species decrease and disappear above 200 ℃. This result implies that K-poisoning impairs the adsorption of NOx species and the reaction might also occur between NH4+ species and NOx species via the L-H mechanism over K-V/Ti.

To further explore the alkali-resistant mechanism, in situ DRIFT spectra of transient reactions were performed at 200 ℃. Firstly, the in situ DRIFT spectra of transient reactions between NO+O2 and pre-adsorbed NH3 were probed for fresh catalysts, as shown in Figs. 4a and b. As for CeSV/Ti (Fig. 4a), the NH4+ (1428 cm−1) [43] and NH3 (1249 cm−1) [44] species appear after the adsorption of NH3, in which the amount of NH4+ species is more than NH3 species. With the introduction of NO+O2, these NH3/NH4+ species gradually decrease within 20 min and a small quantity of nitrates including M-NO2 nitrate species (1346 cm−1) and bridging nitrate (1237 cm−1) emerged [42]. For V/Ti (Fig. 4b), the NH3 species (1233 cm−1) [44] and NH4+ species (1418 cm−1) [45] appear after the adsorption of NH3, where the amount of NH3 species is more than NH4+ species. With the introduction of NO+O2, these NH3/NH4+ species consume gradually within 40 min, and the M-NO2 (1373 cm−1), bridging species (1623 cm−1) and gaseous NO2 (1320 cm−1) increase [42]. It is notable that more NH3/NH4+ species are generated over CeSV/Ti than V/Ti. Besides, the reactivity of NH3/NH4+ species over CeSV/Ti is also higher than that on V/Ti. Figs. 4c and d show the in situ DRIFT spectra of transient reactions for K-poisoned catalysts between NO+O2 and pre-adsorbed NH3 at 200 ℃. As for K-CeSV/Ti (Fig. 4c), the NH3 species (1230 and 1595 cm−1) [38,44] and NH4+ species (1418, 1470 and 1684 cm−1) [36,45,46] appear after the adsorption of NH3. Similar to the fresh CeSV/Ti, the amount of NH4+ species is still more than NH3 species. With the introduction of NO+O2, these NH3/NH4+ species gradually decrease and a small quantity of bidentate nitrates species (1551 cm−1) and M-NO2 species (1340 cm−1) emerge [42]. For K-V/Ti (Fig. 4d), the NH3 species (1217 and 1603 cm−1) [37,40] and NH4+ species (1732 and 1379 cm−1) [43,47] appear after the adsorption of NH3. Meanwhile, the amount of NH3 species is more than NH4+ species. With the introduction of NO+O2, these NH3/NH4+ species gradually decrease while the bidentate nitrates species (1579 cm−1) and M-NO2 species (1359 cm−1) gradually emerge [42]. Clearly, the adsorption of NOx species on V/Ti catalysts is much stronger than CeSV/Ti, indicating that the gaseous NO mainly participates the SCR reaction over CeSV/Ti catalysts.
Figs. 4e and f show in situ DRIFT spectra of the transient reactions at 200 ℃ between NH3 and pre-adsorbed NO+O2 for fresh catalysts. For CeSV/Ti (Fig. 4e), a small quantity of bridging nitrate species (1626 cm−1) and M-NO2 (1381 cm−1) appear after the adsorption of NO+O2 [42]. With the introduction of NH3, these adsorbed NOx species decrease while the NH4+ species (1469 and 1429 cm−1) [36,48] and NH3 species (1250 cm−1) [34] gradually increase. From the above results, it can be inferred that the reaction mainly proceeds between NH4+ species and gaseous NO via the E-R reaction pathway over CeSV/Ti. For V/Ti (Fig. 4f), the bridging nitrates (1628 cm−1), M-NO2 species (1377 cm−1) and adsorbed NO2 (1320 cm−1) appear after the adsorption of NO+O2 [42]. After the introduction of NH3, these NOx species gradually decrease while the NH3 species (1235 cm−1) [44] and NH4+ (1468 and 1410 cm−1) [36,37] increase. Based the above results, the reaction mainly proceeds between NH3 species and NOx species via the L-H reaction pathway over V/Ti.
Figs. 4g and h show the in situ DRIFTS of transient reactions for K poisoned-catalysts between NH3 and pre-adsorbed NO+O2 at 200 ℃. As for K-CeSV/Ti (Fig. 4g), only a small quantity of M-NO2 species (1338 cm−1) are detected after the adsorption of NO+O2 [42]. With introduction of NH3, these NOx species were consumed within 10 min and corresponding NH3 (1324 and 1237 cm−1) [33] and NH4+ (1421 and 1466 cm−1) [39,49] species gradually increase. Therefore, the SCR reaction could still proceed via the reaction between NH4+ species and gaseous NO over K-CeSV/Ti (Fig. 4). For K-V/Ti (Fig. 4h), the bridging nitrates (1601 cm−1), bidentate nitrates (1578 cm−1), M-NO2 (1484 cm−1) and monodentate nitrates (1307 cm−1) species are accumulated after the adsorption of NO+O2 over the K-poisoned V/Ti [42]. However, after the introduction of NH3, these NOx species consumed slowly while the NH3 species (1397 and 1219 cm−1) [40,49] are detected. This result indicates that K-poisoning decreases the reactivity of the adsorbed NOx with NH3 species and blocked the L-H reaction pathway (Fig. 4).
In summary, improved NOx reduction against alkali poisoning over CeSV/Ti catalysts has been originally demonstrated. K poisoned V/Ti shows NOx conversion below 70% while K-poisoned CeSV/Ti shows NOx conversion above 80% from 270 ℃ to 450 ℃. It has been demonstrated that Ce4+–SO42− pair sites play crucial roles in improving the K-resistance of V/Ti catalysts. Here, we propose a deactivation mechanism over K-V/Ti and alkali-resistant mechanism over K-CeSV/Ti catalysts, as seen in Fig. 5. V/Ti catalysts mainly proceed the reaction between NH3 species and NOx species via the L-H reaction pathway. After Ce(SO4)2 modification, the strong interaction between V and Ce sites of Ce4+–SO42− pair leads to the change of L-H reaction pathway into the E-R reaction pathway via the reaction between NH4+ species and gaseous NO. After K-poisoning, the reactivity between the adsorbed NOx species and NH3 species is impaired over K-V/Ti and thus the L-H reaction pathway is blocked. In comparison, K-CeSV/Ti could still proceed the reaction effectively via the E-R reaction pathway because the SO42− sites of Ce4+–SO42− pairs strongly bond with K and protect active sites. This work evidenced an effective strategy to enhance NOx reduction against alkali poisoning over catalysts via constructing Ce4+–SO42− pair sites, contributing to developing alkali-resistant SCR catalysts for practical application in nonelectrical industries.

The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge the National Natural Science Foundation of China (Nos. 22125604, 22106100, 21976117), Shanghai Rising-Star Program (No. 22QA1403700) and Chenguang Program supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (No. 22Z00354).
Supplementary material associated with this article can be found, in the online version, at doi:
A. Richter, J.P. Burrows, H. Nüß, C. Granier, U. Niemeier, Nature 437 (2005) 129–132. doi: 10.1038/nature04092
C. Paolucci, I. Khurana, A.P. Atish, et al., Science 357 (2017) 898–903. doi: 10.1126/science.aan5630
H.K. Chang, G. Qi, K. Dahlberg, W. Li, Science 327 (2010) 1624–1627. doi: 10.1126/science.1184087
C. Paolucci, A.A. Verma, S.A. Bates, et al., Angew. Chem., Int. Ed. 53 (2014) 11828–11833. doi: 10.1002/anie.201407030
G. He, Z. Lian, Y. Yu, et al., Sci. Adv. 4 (2018) eaau4637. doi: 10.1126/sciadv.aau4637
Z. Si, Y. Shen, J. He, et al., Environ. Sci. Technol. 56 (2022) 605–613. doi: 10.1021/acs.est.1c05686
L. Han, S. Cai, M. Gao, et al., Chem. Rev. 119 (2019) 10916–10976. doi: 10.1021/acs.chemrev.9b00202
Y. Zhao, L. Shi, Y. Shen, et al., Environ. Sci. Technol. 56 (2022) 4386–4395. doi: 10.1021/acs.est.1c07996
G. Zhou, P. Maitarad, P. Wang, et al., Environ. Sci. Technol. 54 (2020) 13314–13321. doi: 10.1021/acs.est.0c04536
L. Yan, Y. Ji, P. Wang, et al., Environ. Sci. Technol. 54 (2020) 9132–9141. doi: 10.1021/acs.est.0c03290
S.S.R. Putluru, S.B. Kristensen, J. Due-Hansen, A. Riisager, R. Fehrmann, Catal. Today 184 (2012) 192–196. doi: 10.1016/j.cattod.2011.10.012
S. Gao, P. Wang, X. Chen, et al., Catal. Commun. 43 (2014) 223–226. doi: 10.1016/j.catcom.2013.10.017
J. Due-Hansen, S. Boghosian, A. Kustov, et al., J. Catal. 251 (2007) 459–473. doi: 10.1016/j.jcat.2007.07.016
P. Wang, H. Wang, X. Chen, Z. Wu, ChemCatChem 8 (2016) 787–797. doi: 10.1002/cctc.201501064
P. Wang, S. Gao, H. Wang, et al., Appl. Catal. B 561 (2018) 68–77. doi: 10.1016/j.apcata.2018.05.023
P. Wang, S. Chen, S. Gao, et al., Appl. Catal. B 231 (2018) 299–309. doi: 10.1016/j.apcatb.2018.03.024
P. Zhang, P. Wang, A. Chen, et al., Environ. Sci. Technol. 55 (2021) 11970–11978. doi: 10.1021/acs.est.1c02882
Y. Li, S. Cai, P. Wang, et al., Environ. Sci. Technol. 55 (2021) 9276–9284. doi: 10.1021/acs.est.1c01722
C. Feng, P. Wang, X. Liu, et al., Environ. Sci. Technol. 55 (2021) 11255–11264. doi: 10.1021/acs.est.1c02061
L. Kang, L. Han, J. He, et al., Environ. Sci. Technol. 53 (2019) 938–945. doi: 10.1021/acs.est.8b05637
Z. Chen, S. Ren, M. Wang, et al., Fuel 321 (2022) 124113. doi: 10.1016/j.fuel.2022.124113
H. Zhou, T. Cheng, B. Du, et al., Environ. Sci. Pollut. Res. 29 (2022) 84421–84433. doi: 10.1007/s11356-022-21748-z
L. Wang, J. Zhao, S. Bai, H. Zhao, Z. Zhu, Chem. Eng. J. 254 (2014) 399–409. doi: 10.1016/j.cej.2014.05.096
L. Han, M. Gao, J.Y. Hasegawa, et al., Environ. Sci. Technol. 53 (2019) 6462–6473. doi: 10.1021/acs.est.9b00435
L. Kang, L. Han, P. Wang, et al., Environ. Sci. Technol. 54 (2020) 14066–14075. doi: 10.1021/acs.est.0c05038
Z. Chen, S. Ren, X. Xing, et al., Fuel 335 (2022) 126986.
Z. Chen, S. Ren, M. Wang, Mol. Catal. 531 (2022) 112693. doi: 10.1016/j.mcat.2022.112693
L. Han, M. Gao, C. Feng, L. Shi, D. Zhang, Environ. Sci. Technol. 53 (2019) 5946–5956. doi: 10.1021/acs.est.9b01217
Z. Liu, H. Su, B. Chen, J. Li, S.I. Woo, Chem. Eng. J. 299 (2016) 255–262. doi: 10.1016/j.cej.2016.04.100
Z. Wu, R. Jin, Y. Liu, H. Wang, Catal. Commun. 9 (2008) 2217–2220. doi: 10.1016/j.catcom.2008.05.001
Z. Chen, R. Guo, S. Ren, et al., J. Mater. Chem. A 10 (2022) 21474. doi: 10.1039/d2ta06199f
K. Wang, Z. Gong, H. Luo, et al., Combust. Sci. Technol. 190 (2018) 770–783. doi: 10.1080/00102202.2017.1408598
Y. Zhang, X. Yue, T. Huang, K. Shen, B. Lu, Materials 11 (2018) 1307. doi: 10.3390/ma11081307
Q. Zhang, J. Fan, P. Ning, et al., Appl. Surf. Sci. 435 (2018) 1037–1045. doi: 10.1016/j.apsusc.2017.11.180
J. Liu, J. Meeprasert, S. Namuangruk, et al., J. Phys. Chem. C 121 (2017) 4970–4979. doi: 10.1021/acs.jpcc.6b11175
L. Chen, Z. Si, X. Wu, D. Weng, ACS Appl. Mater. Interfaces 6 (2014) 8134–8145. doi: 10.1021/am5004969
Y. Yu, J. Wang, J. Chen, et al., Ind. Eng. Chem. Res. 53 (2014) 16229–16234. doi: 10.1021/ie502065b
L. Huang, K. Zha, S. Namuangruk, et al., Catal. Sci. Technol. 6 (2016) 8516–8524. doi: 10.1039/C6CY02026G
J. Liu, X. Li, Q. Zhao, et al., Appl. Catal. B 200 (2017) 297–308.
Q. Zhang, H. Wang, P. Ning, et al., Appl. Surf. Sci. 419 (2017) 733–743. doi: 10.1016/j.apsusc.2017.05.056
J. Wang, Z. Yan, L. Liu, et al., Appl. Surf. Sci. 313 (2014) 660–669. doi: 10.1016/j.apsusc.2014.06.043
A. Davydov, Molecular Spectroscopy of Oxide Catalyst Surfaces, John Wiley & Sons Ltd., 2003, Chapter 2, p. 124.
Z. Liu, S. Zhang, J. Li, L. Ma, Appl. Catal. B 144 (2014) 90–95. doi: 10.1016/j.apcatb.2013.06.036
N. Liu, J. Wang, F. Wang, J. Liu, J. Rare Earths 36 (2018) 594–602. doi: 10.1016/j.jre.2017.12.009
H. Hu, S. Cai, H. Li, et al., J. Phys. Chem. C 119 (2015) 22924–22933. doi: 10.1021/acs.jpcc.5b06057
C. Yu, L. Wang, B. Huang, Aerosol Air Qual. Res. 15 (2015) 1017–1027. doi: 10.4209/aaqr.2014.08.0162
X. Weng, X. Dai, Q. Zeng, Y. Liu, Z. Wu, J. Colloid Interface Sci. 461 (2016) 9–14. doi: 10.1016/j.jcis.2015.09.004
S. Wang, R. Guo, W. Pan, et al., Catal. Commun. 89 (2017) 143–147. doi: 10.1016/j.catcom.2016.11.005
K. Liu, F. Liu, L. Xie, W. Shan, H. He, Catal. Sci. Technol. 5 (2015) 2290–2299. doi: 10.1039/C4CY01550A

Figure 1 Plots of NOx conversion versus temperature over CeSV/Ti, K-CeSV/Ti, V/Ti and K-V/Ti catalysts. Reaction conditions: 500 ppm NO, 500 ppm NH3, 5 vol% O2, N2 as the balance gas, and GHSV of 50,000 h−1.

Figure 2 (a) NH3-TPD-MS profiles; (b) H2-TPR profiles; XPS spectra of V 2p (c), O 1s (d), Ce 3d (e) and K 2p (f) of different catalysts.

Figure 3 In situ DRIFTS of NH3 desorption over CeSV/Ti (a), V/Ti (b), K-CeSV/Ti (c), and K-V/Ti (d) catalysts after exposure to NH3 for 1 h at 30 ℃; In situ DRIFTS of NO+O2 desorption over CeSV/Ti (e), V/Ti (f), K-CeSV/Ti (g), and K-V/Ti (h) catalysts after exposure to NO+O2 for 1 h at 30 ℃. The spectra were all recorded at 50, 80, 110, 140, 170, 200, 230, 260 and 300 ℃, respectively.

Figure 4 In situ DRIFTS of the transient reactions between NO+O2 and pre-adsorbed NH3 over CeSV/Ti (a), V/Ti (b), K-CeSV/Ti (c), and K-V/Ti (d) catalysts at 200 ℃ as a function of time; In situ DRIFTS of the transient reactions between NH3 and pre-adsorbed NO+O2 over CeSV/Ti (e), V/Ti (f), K-CeSV/Ti (g), and K-V/Ti (h) catalysts at 200 ℃ as a function of time. The spectra were all recorded at 0, 10, 20, 30, 40, 50, and 60 min, respectively.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
