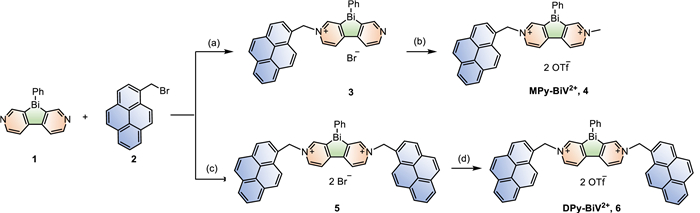
Scheme 1.
The synthetic procedure of pyrene-tethered bismoviologens. Conditions: (a) DCM, r.t. (b) MeOTf, DCM, 0 ℃ to r.t. (c) DMF, 60 ℃, MW. (d) MeOTf, DCM, 0 ℃ to r.t.
Pyrene-tethered bismoviologens for visible light-induced C(sp3)–P and C(sp2)–P bonds formation
Wenqiang Ma , Sikun Zhang , Liang Xu , Bingjie Zhang , Guoping Li , Bin Rao , Mingming Zhang , Gang He

Viologens have achieved extensive applications [1–3] including electron mediators [4,5], supramolecular chemistry [6–8], electrochromism [9,10], electrode materials [11–14], ionic liquid [15–17], due to their good electron-accepting abilities, reversible redox processes, and stable redox states [18]. In particular, viologens can realize continuous multiple switching between dications and radical cations undergoing the reversible single electron transfer [3,18], which makes them as the potential catalysts. Recently, viologens have attracted widespread attention as electron-transfer catalysts (ETC) in the field of organic catalysis and have achieved the preparation of various organic compounds [19–21]. In addition, the exploration of viologens as electron-transfer catalysts in photocatalysis has recently emerged and considerable efforts have been made by scientists. Decorating viologen on the surface of g-C3N4 by intermolecular hydrogen bonds achieved highly effective photocatalytic hydrogen production with a high H2 generation rate and a splendid apparent quantum yield [22]. When coupling viologen with photosensitizer and formate dehydrogenase (FDH), the formic acid was successfully produced via the reduction of CO2, and this method not only contributed to the conversion of CO2 to fuel and organic matter but also provided a strategy for artificial photosystems [23]. Besides, a series of organic reactions, such as the reduction of carbonylic compounds [24], the hydrogenation of C=C and C=O bonds [25], and the selective reduction of α,β-unsaturated compounds [26], were developed by using viologen as an electron media in combination with common photosensitizers, microorganism or enzyme, which promoted the application of viologens in organic photocatalysis.
Although these researchers had reached the goal that viologens were used in the field of photocatalysis and realized the preparation of chemical products, these studies could not avoid using photosensitizers such as noble metal complexes and organic dyes, which not only made reaction complicated but also resulted in environmental problems. Therefore, utilizing viologens as photosensitizers and electron mediators at the same time in the field of photocatalysis had become a promising and feasible alternative. However, it is difficult for viologens to possess good performance of light absorption and rapid photoinduced electron transfer due to viologens' connatural drawbacks, which doubtlessly hinder the application of using viologens as photosensitizers in the field of photocatalysis. Some progress had been made to achieve the purpose. Rigaudy and coworkers reported selective N-demethylation of tertiary amines and cyanation of various alkaloids with N,N-dimethyl-2,7-diazapyrenium (DAP2+) as a photosensitizer that is an analogue of viologens and has better conjugation [27,28]. When chalcogenoviologens that were synthesized by introducing chalcogens into viologen scaffolds were used as photosensitizers, a visible-light-driven hydrogen evolution reaction was reported by our group [29,30]. Recently, our group had developed a visible-light-induced cross-dehydrogenative coupling by using bismoviologens as photosensitizers, which were prepared by the incorporation of bismuth and viologen skeleton [31]. However, the use of viologens as photosensitizers was limited to hydrogen production, the cleavage of C—N bonds, and the construction of C—C bonds. Clearly, expanding photocatalytic reactions using viologens as photosensitizers become urgent that should be settled.
Organophosphorus compounds have received considerable attention as they play an irreplaceable role in pharmaceuticals [32,33], ligands [34–37], biology [38,39] and materials [40,41]. The formation of C-P bonds was mainly through transition metal catalysis [42,43]. In recent years, research in the field of photocatalysis had made great strides and developments [44–49], which have broken new ground for the formation of C–P bonds [50,51]. However, the photosensitizers used in these reactions were still concentrated in precious metals and organic dyes that were either expensive or difficult to modify, which was inconsistent with the concept of green chemistry and sustainable chemistry [52–54]. Compared with common photosensitizers, bismoviologens were easily prepared and modified and had been successfully used in the construction of C—C bonds [31]. The construction of C-P bonds by photocatalysis usually involved the generation of radicals, and bismoviologens could obtain an electron from the reactants under light irradiation to promote the formation of radicals or radical cations. Therefore, it was promising and feasible to use bismoviologens as photosensitizers for the efficient construction of C-P bonds. Because, pyrene exhibits a long luminescence lifetime and strong light absorption, and it has been widely used to enhance the light absorption performance in molecule design [55–57]. Therefore, bonding pyrene with bismoviologens skeleton would further improve the opto-electronic properties of bismoviologens and enhance the efficiency of photocatalytic reactions.
Based on the considerations, two pyrene-tethered bismoviologens were synthesized, and the opto-electronic properties of the two molecules were fine-tuned by the number of substituents. More importantly, they exhibited stronger performance of light absorption and faster photoinduced electron transfer process, thus they were used as photosensitizers to achieve C(sp3)–P and C(sp2)–P bonds formation under visible-light photocatalysis with high efficiency.
To obtain the desired materials required for photocatalysis, the pyrene-tethered bismoviologens (Py-BiV2+) were prepared through the reaction of dipyridinobismole (1) with 1-bromomethylpyrene (2) (Scheme 1) [58]. Firstly, mono-pyrene substituted bismoviologen (MPy-BiV2+,4) was synthesized by the reaction of 1 (1 equiv.) and 2 (0.3 equiv.), followed by the methylation and anion exchange simultaneously with excess methyl triflate (MeOTf). Then, the reaction started from between 1 (1 equiv.) and 2 (6 equiv.), subsequently exchange anion to OTf with excess methyl triflate (MeOTf), affording di-pyrene-substituted bismoviologen (DPy-BiV2+,6). The thermal properties of Py-BiV2+ were examined by thermogravimetric analysis (TGA), which showed a slow weight loss up to 200 ℃ and a rapid weight loss starting at approximately 300 ℃, indicating that Py-BiV2+ were stable below 200 ℃ (Fig. S1 in Supporting information).
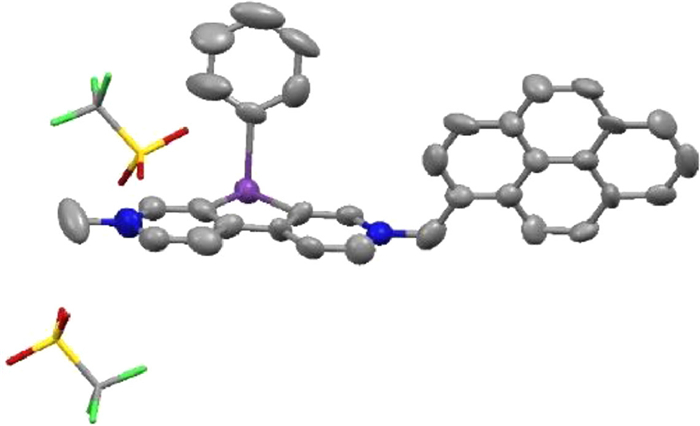
The solid structure of MPy-BiV2+ (4) was verified by single-crystal X-ray diffraction analysis. As shown in Fig. 1, tricyclic units exhibited almost complete planarity in the single-crystal structure of 4 and the phenyl on the bismuth atom was nearly perpendicular to the tricyclic units, which was in agreement with the previous report [31]. The packing in 4 showed two different π−π stacking interactions. The distance between the pyrene group and tricyclic units of adjacent molecules is 3.441 Å, which indicated π−π stacking interactions, whereas the two pyrene groups of neighboring molecules showed a weaker π−π stacking interaction with a distance of 3.469 Å (Fig. S2 in Supporting information).
With the desired molecules in hand, the UV–vis absorption spectra were collected in acetonitrile to investigate the optical properties. As shown in Fig. 2a, Py-BiV2+ displayed pronounced absorptions peaks close to 350 nm with molar absorption coefficients (ε) of 2.0 × 104 L mol−1 cm−1 (4) and 2.1 × 104 L mol−1 cm−1 (6), respectively. A distinct shoulder was observed at 273 nm for 4, 286 nm for 6. their molar absorption coefficients are 1.8 × 104 L mol−1 cm−1 (4) and 1.6 × 104 L mol−1 cm−1 (6). The result of absorption spectra led to a yellow color that is visible to the naked eye. Besides, there was a weak absorption, which extended to about 450 nm. Compared with dimethyl bismoviologen (Me-BiV), 4 and 6 showed a similar red-shifted absorption and a reduced energy gap. This phenomenon stemmed from the elevation of HOMO energy levels since HOMO was located on the electron-rich pyrene units. The photoluminescence spectra measure of pyrene, Me-BiV, and Py-BiV2+ ensued (Fig. 2b). When the excitation wavelength is 340 nm, pyrene exhibited two characteristic PL emission bands centered at 375 nm and 390 nm, whereas Py-BiV2+ had virtually no emission peaks in these two regions. This result indicated that the luminescence of the pyrene group in Py-BiV2+ was quenched due to the accelerated photoinduced electron transfer (PET) between pyrene and bismoviologen skeleton [59,60]. A similar phenomenon was observed at the excitation wavelength of 365 nm.

As viologen derivatives, Py-BiV2+ also showed good electron-accepting ability. The acetonitrile solution of 4 and N,N-diisopropylethylamine (DIPEA, electron donor) exhibited obvious absorption peaks at ca. 400 nm and ca. 630 nm under white LED irradiation, which implied the production of radical cations (4′) (Fig. S3a in Supporting information). In comparison, the solution of Me-BiV and DIPEA showed a very small change in UV–vis absorption spectra (Fig. S3b in Supporting information). The increase in absorption strength at ca. 630 nm of 4 is 2.5 times that of Me-BiV2+. This result demonstrated that the photoinduced electron transfer between 4 and DIPEA is much faster, because the strong light absorption of the pyrene group promotes the photoinduced electron transfer (Scheme S1 in Supporting information). Since the absorption of the pyrene group is mainly in the ultraviolet region, the absorption intensity of the 4 and DIPEA solution at 630 nm was significantly enhanced after UV irradiation, which meant more radical cations (4′) were generated (Fig. 2c). The increase in absorption strength at ca. 630 nm of 4 becomes 4 times that of Me-BiV (Fig. S4 in Supporting information). The results suggested a more efficient photoinduced electron transfer and further confirmed that 4 is easier to accept the electron than Me-BiV. The advantage laid a foundation for 4 as a photosensitizer.
Py-BiV2+ could also be used as an electrochromic material. The electrochromic devices (ECDs) consisting of two pieces of indium tin oxide (ITO) and the DMF solution of Py-BiV2+ without any electrolytes were constructed. When adjusting the applied potential to −0.7 V, 4-based ECD exhibited a color change from yellow to blue-green (Fig. 2d), and in the case of 6-based ECD, a color change had appeared from orange to blue-green (Fig. S6 in Supporting information), which reflected that the dications (4 and 6) were gradually reduced to radical cations (4′ and 6′, Supporting information). When the applied potential was increased from −0.7 V to −1.0 V, the blue-green color of 4 or 6-based ECD faded away because of a decrease of radical cations (4′ and 6′) (Fig. 2d and Fig. S6 in Supporting information). Subsequently, in situ spectroelectrochemistry experiments based on ECDs were carried out to investigate the variation of the absorption spectra of Py-BiV2+ with the time of externally applied voltages. As shown in Fig. 2e, 4-based ECD was gradually reduced and two distinct absorption peaks at 581 nm and 634 nm kept on rising accompanied by a weak absorption peak at 795 nm with a voltage of −0.7 V. By comparison, 6-based ECD displayed the continuously increasing absorption peak at 596 nm and 637 nm, and no significant absorption intensity was observed in the near-IR region at −0.7 V (Fig. S6). When the applied potential was increased to −1.0 V, the absorption intensity of 4/6-based ECDs in the visible region decreased to a certain extent (Fig. 2f and Fig. S6). The phenomenon was associated with the reduction of the radical cations. The radical stability was tested, and it could be found that the radical stability of 4-based ECD is four times higher than that of Me-BiV (Fig. S7 in Supporting information).
Meanwhile, the interactions of Py-BiV2+ with the appropriate chemical reagents led to additional two redox states, which exhibited significantly different colors compared with the parent due to differences in absorption spectra. Accordingly, the UV–vis absorption spectra of all species under argon were obtained. The addition of zinc powder to the DMF solution of 4 and 6 resulted in the conversion of dications to the radical cations (4′ and 6′), and a conspicuous absorption band was detected in the range of 500 nm-800 nm accompanied by a color change from yellow or orange to blue-green respectively (Fig. 2g and Fig. S7). The radical cations (4′ and 6′) were confirmed by electron paramagnetic resonance (EPR) spectra (Fig. S11 in Supporting information). The further treatment of radical cations (4′ and 6′) with Na resulted in yellow neutral species (4′′ and 6′′), which showed a broad and attenuated absorption band in the visible region (Fig. 2g and Fig. S8 in Supporting information). The results suggested that the phenomenon of chemical reductions was consistent with that of electrochemical results. Besides, the emission spectra of these reduced species were further collected. As expected, radical cations (4′ and 6′) and neutral species (4′′ and 6′′) showed significant photoluminescent properties (Fig. S9 in Supporting information).
The measure of cyclic voltammetry in dimethylformamide (DMF) was carried out to investigate the electrochemical properties of Py-BiV2+ and the results were summarized in Table S2 (Supporting information). The cyclic voltammograms of two Py-BiV2+ showed two pairs of redox peaks at a sequence of scanning speeds ranging from 10 mV/s to 1000 mV/s, which is ascribed to two reversible redox reactions that were the innate nature of viologens (Fig. S10 in Supporting information). Compared to previous studies, the first reduction of the 4 and 6 was more positive than that of Me-BiV. This result implied that Py-BiV2+ are more capable of accepting electrons than Me-BiV. Although the reduction potential of 4 is slightly more negative than that of dibenzyl bismoviologen, its stronger light absorption performance and more red-shifted absorption make it favorable for photocatalysis.
To gain deeper insights into the experimental results for Py-BiV2+, the density functional theory (DFT) calculations were conducted. The distributions of the HOMO and LUMO were depicted, the HOMO distributions for 4 and 6 were similar, which were situated on the pyrene units, whereas the LUMO distributions were mainly situated on the bismoviologen skeleton (Fig. 3, Figs. S21 and S22 in Supporting information). The slightly lower LUMO energy levels of Py-BiV2+ than Me-BiV suggested that the reduction potential of Py-BiV2+ was slightly more positive than that of Me-BiV, which was in agreement with their reduction potential obtained by the measure of cyclic voltammetry. However, their HOMO energy levels were higher than Me-BiV which led to a more narrow energy gap, these results confirmed the red-shifted absorption band in both molecules. Predictably, the UV–vis absorption of theoretical calculations basically coincided with experimental data (Figs. S15-S20 in Supporting information). In the case of 4, The HOMO → LUMO+2 transition was mainly responsible for the maximum absorption (Table S7 in Supporting information). The maximum absorption of 6 principally originated from the HOMO-1 → LUMO+2 transition and the HOMO → LUMO+3 transition (Table S8 in Supporting information). The energy gaps that were separately calculated from the HOMO → LUMO+2, HOMO-1 → LUMO+2, and HOMO → LUMO+3 energy levels were similar for 4 and 6 (Fig. 3), in agreement with the gained results of energy gaps from UV–vis absorption.
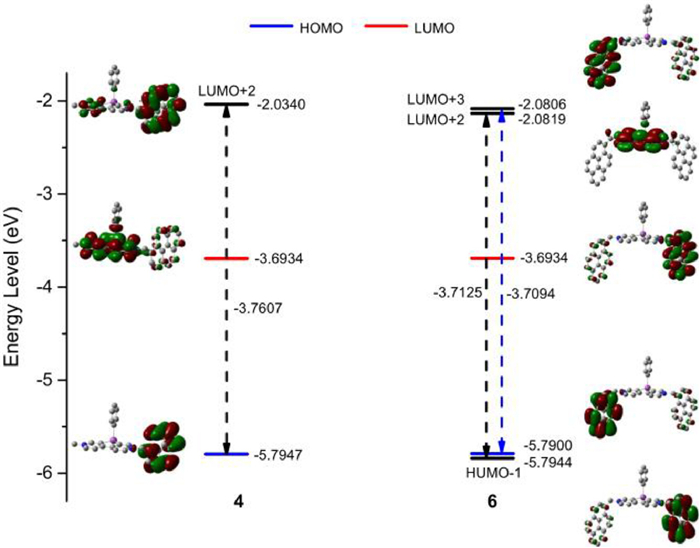
In addition, the electrostatic potential maps were presented in different colours (Fig. S24 in Supporting information). For the dications, the more positive electrostatic potential was observed on three-membered heterocycles, relative to the radical cations, suggesting that three-membered heterocycles served as the region of accepting electrons when interacting with other compounds. The charge distribution was separately calculated using natural bond orbital (NBO) charges and Mulliken charges, which could be distinguished directly by the color of the spheroids (Figs. S25 and S26 in Supporting information). The most negatively and the most positively charged were indicated as the reddest and the greenest spheroids, so the most positive atom was the bismuth atom, the negative charge was mainly distributed on nitrogen atoms and carbon atoms that were directly connected to the bismuth atom.
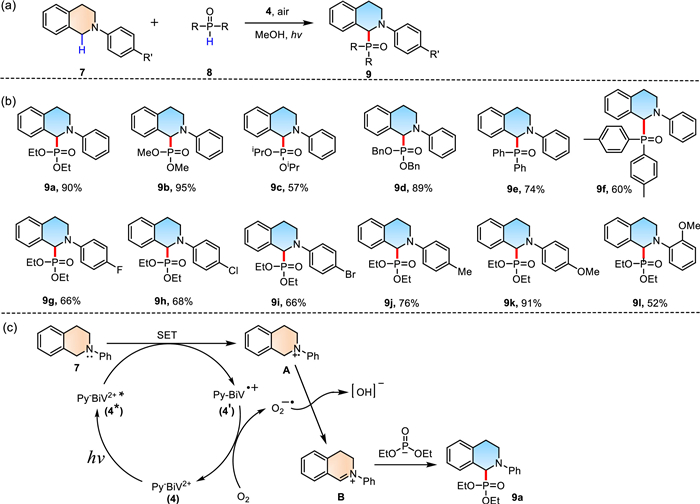
The phosphorus-containing compounds are an essential organic matter that has extensive application and their synthesis has aroused increasing attention. Constructing C-P bonds by photoredox catalysis is considered a green and environmentally friendly synthetic strategy [61,62]. To the best of our knowledge, the construction of C-P bonds using viologen derivatives as photosensitizers has not been reported. Considering the previous research of bismoviologens and the characteristics of Py-BiV2+ [31], an aerobic reaction of C(sp3)-P bond formation was carried out in the air. Diethyl phosphite (8a) was selected as a model substrate for reaction with N-phenyltetrahydroisoquinoline (7a), and 4 was used as a photosensitizer and electron media (Scheme 2). The initial investigation focused on the effect of solvents (Table S3, entries 1–7). The experiment results revealed that the target product was formed in moderate yield, when CH3CN, DMF, EtOH, THF, and acetone were employed as solvents (entries 1–5). The reaction was carried out in CH3OH to provide the desired product (9a) in excellent yields (90% yield, entry 7). In addition, H2O was used as the solvent and obtained 32% yields of 9a (entry 6). Therefore, CH3OH was chosen as a solvent to investigate the effects of other conditions. Lowering the amount of catalyst to 1 mol% decreased the yield of 9a to 82% (entry 8). Subsequently, a series of control experiments were carried out. As expected, trace product was observed without photosensitizers or light (entries 10–11), and a little of the product (14% yield) was detected in argon (entries 9). The results showed that the use of 4 as a photosensitizer was required. Replacing photosensitizer 4 with other viologen derivatives led to a decrease in the yield of 9a (entries 12–14). Furthermore, the reaction of different phosphite or phosphine oxides with N-aryltetrahydroisoquinoline (7) could gain the desired products (9b-9l) in moderate to good yields (52%–95%) (Scheme 2b).
The thermodynamic feasibility of the photoinduced single electron transfer (SET) was analyzed by the oxidation–reduction potentials. The exited state reduction potential of E(4*/4′) was calculated to be +2.15 V (vs. SCE) (see Supporting information for details). The oxidation potential of the N-phenyltetrahydroisoquinoline (7a) was +0.82 V (vs. SCE) [61]. Therefore, the single electron transfer (SET) between N-phenyltetrahydroisoquinoline (7a) and the excited 4 can occur smoothly. Next, the luminescent quenching experiments of 4 were conducted with substrate N-phenyltetrahydroisoquinoline (7a), leading to an obvious quenching phenomenon (Figs. S13 and S14 in Supporting information).
The plausible mechanism of C(sp3)-P bond formation based on the experimental results and related literature reports is depicted in Scheme 2c [61,63–65]. Upon excitation of 4, a single electron transfer from 7 to 4 * occurred with the generation of the radical cation A and the radical cation 4′. The radical cation 4′ was oxidized by O2 and regenerated ground state 4. Meanwhile, radical cation A lost a hydrogen atom by a radical anion to generate iminium ion B, followed by trapping with nucleophiles to give the desired product 9.
It is noting that the construction of C(sp2)–P bond is not a negligible part of the preparation of organophosphorus compounds, and considerable efforts have gone into constructing the C(sp2)–P bonds [66–68]. Photocatalytic dehydrogenation coupling reactions provide a green and efficient method of C(sp2)–P bond formation avoiding pre-functionalization of reactants [69,70].
Inspired by previous research [69,71], we started an investigation for photocatalytic aerobic C(sp2)–P bond formation of benzothiazole derivatives with diphenylphosphine oxide with 4 as a photosensitizer (Scheme 3). Firstly, the reaction conditions were optimized for the direct construction of the C(sp2)–P bond. Various solvents were examined in the air at room temperature (Table S4, entries 1–6). When H2O was used as the solvent, the desired product was provided in moderate yield (63%, entry 6). The product was gained in good yield when increasing the amount of diphenylphosphine oxide to 6 equiv. (88%, entry 7). Relevant control reactions indicated that light, O2, and photosensitizer 4 are indispensable for this reaction (entries 8–10). Different candidates of viologens were examined, and investigations revealed that 4 was the best photosensitizer for this transformation (entries 11–13). With the optimal reaction conditions in hand, then the substrate scope of this cross-coupling reaction was evaluated. As summarized in Scheme 3b, a variety of benzothiazole derivatives and diarylphosphine reacted smoothly to produce corresponding C(sp2)–P coupled products with good conversion yields (56%–88%).
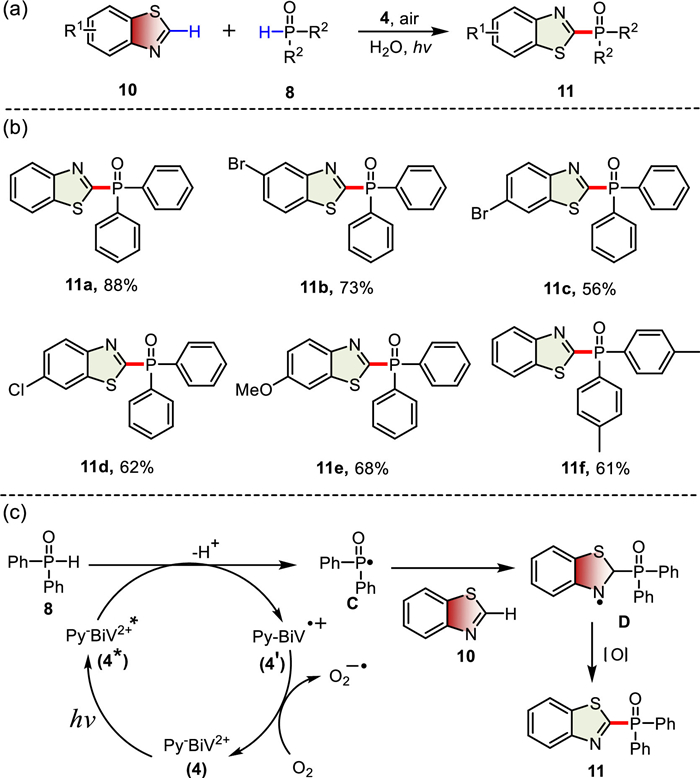
A plausible mechanism for the construction of C(sp2)–P bond reaction is also proposed [71]. The excited state 4* interacted with diphenylphosphine oxide 8, followed by deprotonation to access the radical species C and radical cation 4′.Then, the radical cation 4′ was oxidized by O2 and regenerated ground state 4 to complete the photocatalytic cycle. Meanwhile, radical species C attacked benzothiazole 10 to furnish radical intermediate D, which underwent rearomatization by oxidation and deprotonation to obtain the final product 11 (Scheme 3c). Notably, this is the first time that viologen derivatives were used as photosensitizers for visible-light-induced C-P bond formation. The results demonstrated that viologen derivatives have a wide application prospect in organic photocatalysis.
In summary, we synthesized two bismoviologen derivatives (Py-BiV2+) containing different numbers of pyrene groups. Adopting diversified characterization methods investigated their optical and electrochemical properties, which could be adjusted by the pyrene group with good optical properties. The characterization not only demonstrated the effect of the pyrene group, but also revealed that Py-BiV2+ had strong light absorption performance and reversible redox processes. Py-BiV2+ exhibited a faster and easier photoinduced electron transfer process, and the generated 4′ (radical cation) showed 4 times more stability than Me-BiV. Py-BiV2+ were used as electrochromic materials and the corresponding electrochromic devices had been assembled to exhibit multi-color displays. Simultaneously, Py-BiV2+ could be used as photosensitizers and exhibited excellent catalytic activity that had yields of up to 95% for the construction of C(sp3)–P and yields of up to 88% for the construction of C(sp2)–P bonds under visible light, respectively. These results demonstrated that the Py-BiV2+ are a class of promising functional materials and promoted the development of viologen derivatives and their application as photosensitizers in photocatalysis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Natural Science Foundation of China (Nos. 22175138, 21875180), the Key Research and Development Program of Shaanxi (No. 2021GXLH-Z023), the Independent Innovation Capability Improvement Project of Xi'an Jiaotong University (No. PY3A066). We thank Dr. Lu Bai and Dr. Yu Wang at the Instrument Analysis Center of Xi'an Jiaotong University for HRMS, and photoluminescence measurements. We thank Lijing Ma at State Key Laboratory of Multiphase Flow in Power Engineering of Xi'an Jiaotong University for her assistance with EPR measurements.
Supplementary material associated with this article can be found, in the online version, at doi:
T.P. Nguyen, A.D. Easley, N. Kang, et al., Nature 593 (2021) 61–66. doi: 10.1038/s41586-021-03399-1
L. Liu, Q. Liu, R. Li, M.S. Wang, G.C. Guo, J. Am. Chem. Soc. 143 (2021) 2232–2238. doi: 10.1021/jacs.0c10183
J. Ding, C. Zheng, L. Wang, et al., J. Mater. Chem. A 7 (2019) 23337–23360. doi: 10.1039/C9TA01724K
H. Chen, V. Brasiliense, J. Mo, et al., J. Am. Chem. Soc. 143 (2021) 2886–2895. doi: 10.1021/jacs.0c12664
K. Sokolowski, J. Huang, T. Foldes, et al., Nat. Nanotechnol. 16 (2021) 1121–1129. doi: 10.1038/s41565-021-00949-6
L. Chen, Y.C. Zhang, W.K. Wang, et al., Chin. Chem. Lett. 26 (2015) 811–816. doi: 10.1016/j.cclet.2015.01.036
Q. Ji, L. Fan, S. Liu, et al., Chin. Chem. Lett. 32 (2021) 3998–4001. doi: 10.1016/j.cclet.2021.05.036
A. Swartjes, P.B. White, M. Lammertink, J. Elemans, R.J.M. Nolte, Angew. Chem. Int. Ed. 60 (2021) 1254–1262. doi: 10.1002/anie.202010335
D.G. Seo, H.C. Moon, Adv. Funct. Mater. 28 (2018) 1706948. doi: 10.1002/adfm.201706948
H. Ling, J. Wu, F. Su, Y. Tian, Y.J. Liu, Nat. Commun. 12 (2021) 1010. doi: 10.1038/s41467-021-21086-7
S. Jin, E.M. Fell, L. Vina-Lopez, et al., Adv. Energy Mater. 10 (2020) 2000100. doi: 10.1002/aenm.202000100
W. Wu, J. Luo, F. Wang, B. Yuan, T.L. Liu, ACS Energy Lett. 6 (2021) 2891–2897. doi: 10.1021/acsenergylett.1c01146
C.R. Bridges, M. Stolar, T. Baumgartner, Batteries Supercaps 3 (2020) 268–274. doi: 10.1002/batt.201900164
H. Fan, K. Li, X. Liu, et al., ACS Appl. Mater. Interfaces 12 (2020) 28451–28460. doi: 10.1021/acsami.0c09589
J.W. Kim, J.M. Myoung, Adv. Funct. Mater. 29 (2019) 1808911. doi: 10.1002/adfm.201808911
B. Gelinas, D. Das, D. Rochefort, ACS Appl. Mater. Interfaces 9 (2017) 28726–28736. doi: 10.1021/acsami.7b04427
H. Tahara, Y. Tanaka, S. Yamamoto, et al., Chem. Sci. 12 (2021) 4872–4882. doi: 10.1039/D0SC06244H
L. Striepe, T. Baumgartner, Chem. Eur. J. 23 (2017) 16924–16940. doi: 10.1002/chem.201703348
T. Kitazume, T. Ikeya, J. Org. Chem. 53 (1988) 2349–2350. doi: 10.1021/jo00245a040
L. Coche, B. Ehui, D. Limosin, J.C. Moutet, J. Org. Chem. 55 (1990) 5905–5910. doi: 10.1021/jo00310a025
J.K. Tang, S.B. Yu, C.Z. Liu, et al., Asian J. Org. Chem. 8 (2019) 1912–1918. doi: 10.1002/ajoc.201900435
Y.N. Liu, C.C. Shen, N. Jiang, et al., ACS Catal. 7 (2017) 8228–8234. doi: 10.1021/acscatal.7b03266
T. Noji, T. Jin, M. Nango, N. Kamiya, Y. Amao, ACS Appl. Mater. Interfaces 9 (2017) 3260–3265. doi: 10.1021/acsami.6b12744
J.R. Herance, B. Ferrer, J.L. Bourdelande, J. Marquet, H. Garcia, Chem. Eur. J. 12 (2006) 3890–3895. doi: 10.1002/chem.200501365
S.F. Rowe, G. Le Gall, E.V. Ainsworth, et al., ACS Catal. 7 (2017) 7558–7566. doi: 10.1021/acscatal.7b02736
M.K. Peers, H.S. Toogood, D.J. Heyes, et al., Catal. Sci. Technol. 6 (2016) 169–177. doi: 10.1039/C5CY01642H
J. Santamaria, R. Ouchabane, J. Rigaudy, Tetrahedron Lett. 30 (1989) 2927–2928. doi: 10.1016/S0040-4039(00)99160-3
J. Santamaria, M.T. Kaddachi, J. Rigaudy, Tetrahedron Lett. 31 (1990) 4735–4738. doi: 10.1016/S0040-4039(00)97719-0
G. Li, L. Xu, W. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 4897–4901. doi: 10.1002/anie.201711761
G. Li, R. Song, W. Ma, et al., J. Mater. Chem. A 8 (2020) 12278–12284. doi: 10.1039/D0TA02930K
W. Ma, L. Xu, S. Zhang, et al., J. Am. Chem. Soc. 143 (2021) 1590–1597. doi: 10.1021/jacs.0c12015
L. Weinschenk, D. Schols, J. Balzarini, C. Meier, J. Med. Chem. 58 (2015) 6114–6130. doi: 10.1021/acs.jmedchem.5b00737
W. Feng, X.Y. Teo, W. Novera, et al., J. Med. Chem. 58 (2015) 6456–6480. doi: 10.1021/acs.jmedchem.5b00848
F. Zhao, L. Xin, Y. Zhang, X. Jia, Chin. Chem. Lett. 29 (2018) 493–496. doi: 10.1016/j.cclet.2017.08.004
X. Wang, Z. Han, Z. Wang, K. Ding, Acc. Chem. Res. 54 (2021) 668–684. doi: 10.1021/acs.accounts.0c00697
D. Xiao, X. Zhao, J. Lei, M. Zhu, L. Xu, Chin. Chem. Lett. 32 (2021) 3031–3033. doi: 10.1016/j.cclet.2021.03.068
D. Wang, Z. Song, J. Zhang, T. Xu, Org. Chem. Front. 8 (2021) 1125–1131. doi: 10.1039/D0QO01399D
P. Kumar, M.H. Caruthers, Acc. Chem. Res. 53 (2020) 2152–2166. doi: 10.1021/acs.accounts.0c00078
Y. Sun, L. Cui, Q. Li, et al., Chin. Chem. Lett. 33 (2022) 516–518. doi: 10.1016/j.cclet.2021.06.050
C. Queffelec, M. Petit, P. Janvier, D.A. Knight, B. Bujoli, Chem. Rev. 112 (2012) 3777–3807. doi: 10.1021/cr2004212
T. Baumgartner, Acc. Chem. Res. 47 (2014) 1613–1622. doi: 10.1021/ar500084b
C.S. Demmer, N. Krogsgaard-Larsen, L. Bunch, Chem. Rev. 111 (2011) 7981–8006. doi: 10.1021/cr2002646
Z. Yang, J.J. Wang, Angew. Chem. Int. Ed. 60 (2021) 27288–27292. doi: 10.1002/anie.202112285
K.L. Skubi, T.R. Blum, T.P. Yoon, Chem. Rev. 116 (2016) 10035–10074. doi: 10.1021/acs.chemrev.6b00018
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075–10166. doi: 10.1021/acs.chemrev.6b00057
D.P. Hari, T. Hering, B. Konig, Angew. Chem. Int. Ed. 53 (2014) 725–728. doi: 10.1002/anie.201307051
H. Bartling, A. Eisenhofer, B. König, R.M. Gschwind, J. Am. Chem. Soc. 138 (2016) 11860–11871. doi: 10.1021/jacs.6b06658
Y. Wang, B. Qiu, L. Hu, G. Lu, T. Xu, ACS Catal. 11 (2021) 9136–9142. doi: 10.1021/acscatal.1c02480
X. Wang, F. Liu, T. Xu, Chin. Chem. Lett. (2022) 10.16/j.cclet.2022.06.047. doi: 10.16/j.cclet.2022.06.047
B.G. Cai, J. Xuan, W.J. Xiao, Sci. Bull. 64 (2019) 337–350. doi: 10.1016/j.scib.2019.02.002
S.P.M. Ung, V.A. Mechrouk, C.J. Li, Synthesis 53 (2021) 1003–1022. doi: 10.1055/s-0040-1705978
M. Rueping, S. Zhu, R.M. Koenigs, Chem. Commun. 47 (2011) 8679–8681. doi: 10.1039/c1cc12907d
W.P. To, Y. Liu, T.C. Lau, C.M. Che, Chem. Eur. J. 19 (2013) 5654–5664. doi: 10.1002/chem.201203774
Y. He, H. Wu, F.D. Toste, Chem. Sci. 6 (2015) 1194–1198. doi: 10.1039/C4SC03092C
T.M. Figueira-Duarte, K. Mullen, Chem. Rev. 111 (2011) 7260–7314. doi: 10.1021/cr100428a
J. Zhao, W. Wu, J. Sun, S. Guo, Chem. Soc. Rev. 42 (2013) 5323–5351. doi: 10.1039/c3cs35531d
C. Wang, T. Jiang, X. Ma, Chin. Chem. Lett. 31 (2020) 2921–2924. doi: 10.1016/j.cclet.2020.03.021
S.Y. Park, J.H. Yoon, C.S. Hong, et al., J. Org. Chem. 73 (2008) 8212–8218. doi: 10.1021/jo8012918
Z. Gong, J. Bao, K. Nagai, et al., J. Phys. Chem. B 120 (2016) 4286–4295. doi: 10.1021/acs.jpcb.6b01844
P. Jagadesan, S.R. Valandro, K.S. Schanze, Mater. Chem. Front. 4 (2020) 3649–3659. doi: 10.1039/D0QM00476F
D.P. Hari, B. König, Org. Lett. 13 (2011) 3852–3855. doi: 10.1021/ol201376v
K. Luo, W.C. Yang, L. Wu, Asian J. Org. Chem. 6 (2017) 350–367. doi: 10.1002/ajoc.201600512
J.J. Zhong, Q.Y. Meng, G.X. Wang, et al., Chem. Eur. J. 19 (2013) 6443–6450. doi: 10.1002/chem.201204572
X.Z. Wang, Q.Y. Meng, J.J. Zhong, et al., Chem. Commun. 51 (2015) 11256–11259. doi: 10.1039/C5CC03421C
Y. Zhi, S. Ma, H. Xia, et al., Appl. Catal. B: Environ. 244 (2019) 36–44. doi: 10.1016/j.apcatb.2018.11.032
H.J. Zhang, W. Lin, Z. Wu, W. Ruan, T.B. Wen, Chem. Commun. 51 (2015) 3450–3453. doi: 10.1039/C4CC10017D
L. Li, J.J. Wang, G.W. Wang, J. Org. Chem. 81 (2016) 5433–5439. doi: 10.1021/acs.joc.6b00786
L. Chen, X.Y. Liu, Y.X. Zou, Adv. Synth. Catal. 362 (2020) 1724–1818. doi: 10.1002/adsc.201901540
K. Luo, Y.Z. Chen, W.C. Yang, J. Zhu, L. Wu, Org. Lett. 18 (2016) 452–455. doi: 10.1021/acs.orglett.5b03497
Z. Zhang, Z. Zhao, Y. Hou, et al., Angew. Chem. Int. Ed. 58 (2019) 8862–8866. doi: 10.1002/anie.201904407
P. Peng, L. Peng, G. Wang, et al., Org. Chem. Front. 3 (2016) 749–752. doi: 10.1039/C6QO00049E

Scheme 1 The synthetic procedure of pyrene-tethered bismoviologens. Conditions: (a) DCM, r.t. (b) MeOTf, DCM, 0 ℃ to r.t. (c) DMF, 60 ℃, MW. (d) MeOTf, DCM, 0 ℃ to r.t.

Figure 1 X-ray single-crystal structure of 4 (thermal ellipsoids set at 50% probability, H atoms are omitted for clarity).

Figure 2 (a) UV–vis spectra of Py-BiV2+ in CH3CN, c ≈ 6.5 × 10−5 mol/L; extrapolated band gaps (Eg) and photographs of Py-BiV2+ are shown as insets. (b) Photoluminescence spectra of pyrene, Me-BiV, 4 and 6 in CH3CN under ambient conditions (Ex = 340 nm), photoluminescence spectra (insets) of pyrene, Me-BiV, 4 and 6 in CH3CN under ambient conditions (Ex = 365 nm). (c) UV–vis spectra changes of 4 irradiated with UV in the presence of DIPEA under Ar atmosphere. (d) Solution-based ECD with 4 as an electrochromic material. Spectroelectrochemistry of 4 : (e) first reduction, (f) second reduction. (g) UV–vis spectra of radical cation (4′) and neutral species (4′′).

Scheme 2 (a) Visible-light-induced C(sp3)–P bonds formation catalyzed by Py-BiV2+ . (b) Substrate scope of the C(sp3)–P bonds formation reaction. (c) Proposed mechanism of the C(sp3)–P bonds formation reaction.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

