State Key Lab of Fine Chemicals, School of Chemical Engineering, Liaoning Key Lab for Energy Materials and Chemical Engineering, Dalian University of Technology, Dalian 116024, China
b.
College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China
* Corresponding authors at: State Key Lab of Fine Chemicals
School of Chemical Engineering
Liaoning Key Lab for Energy Materials and Chemical Engineering
Received Date:
26 June 2022 Accepted Date:
20 October 2022 Revised Date:
09 October 2022 Available Online:
15 June 2023
Abstract:
The water promotion effects, where water can provide a solution-mediated reaction pathway in various heterogeneous chemical catalysis, have been presented and attracted wide attention recently, yet, the rational design of catalysts with a certain ability of enhancing water-induced reaction process is full of challenges and difficulties. Here, we show that by incorporating alkali (Na, K) cations as an electronic and/or structural promoter into Pd/rGO-ZnCr2O4 (rGO, reduced graphene oxide), the obtained Pd(Na)/rGO-ZnCr2O4 as a representative example demonstrates an outstanding benzyl alcohol oxidation activity in the Pickering emulsion system in comparison to the alkali-free counterpart. The response experiments of water injection confirm the enhanced activity, and the Na-modified catalyst can further enhance the promotion effects of water on the reaction. The effects of alkali cations for Pd nanoparticles are identified and deciphered by a series of experimental characterizations (XPS, in situ CO-DRIFTS, and CO-TPR coupled with MS), showing that there is abundant −OH on the surface of the catalyst, which is stabilized by the formation of Pd−OHx. The alkali-stabilized Pd−OHx is helpful to enhance the water-induced reaction process. According to the results of in situ Raman as well as UV-vis absorption spectra, the Na-modulated Pd(Na)/rGO-ZnCr2O4 enables the beneficial characteristics for distorting the benzyl alcohol structure and enhancing the adsorption of benzyl alcohol. Further, the mechanism for enhanced water promotion effects is rationally proposed. The strategy of alkali cations-modified catalysts can provide a new direction to effectively enhance the chemical reaction involving small molecule water.
Pickering emulsion, acting as an emerging heterogeneous catalysis system, can provide a particular solvent environment, where chemical reactions are driven by the emulsifier with active sites adsorbed at the interface of two-phase generally made of water and organic solvent [1–3]. The unique emulsion catalysis properties enable regulating the selectivity of chemical reactions according to the relative solubility of reactants and products [1,4,5]. In addition, the selectivity and reactivity of chemical reactions can also be adjusted by the controlled active sites on the emulsion catalysts on the side of the water or oil [6]. Recently, our previous work confirmed that the water was one of the solvents participated in the oxidation reaction of alcohol, and it was found that the activity of water molecules was the key to promote the whole reaction efficiency [7,8]. Therefore, it is extremely desirable to rationally design the catalysts with a certain ability of enhancing the water-induced reaction process, which is also full of difficulties and challenges.
It has been well proved that alkali as an electron donor and/or structural regulator can modify the metal-supported catalyst with high catalytic performance compared with the alkali-free catalyst [9,10]. Up to now, the promotion effects of alkali have been widely explored and applied in various heterogeneous reactions, such as water-gas shift reaction [11–14], Fischer-Tropsch synthesis [15–17], selective hydrogenation [9,10,18,19], CO oxidation [20], selective oxidation [21–26], and organic reaction [22,25,27]. A prevailing view is that the alkali metal can modulate the oxidation states and coordination environments of the active sites (Pd [28], Pt [12–14,20], Au [12,22], Ru [9,19], Ir [26]) and simultaneously promote the dispersion of metal nanoparticles on the carrier. Yet, the potential structure-activity relationships between the catalytic activity and the critical role of alkali species are still not clear. This is finally detrimental to the development and practical application for the strategy of alkali-modulated catalysts [29].
Here, we recognize and decipher the effects of alkali cations on Pd catalyst for selective oxidation of benzyl alcohol (BA) to benzaldehyde (BAD), a chemical reaction system involving in water-promoted effects. The Pd(Na)/rGO-ZnCr2O4 catalyst with Na cations constructed by the coprecipitation-calcination method exhibits a turnover frequency (TOF) value of 469 h−1 and the apparently low activation energy (Ea) of 45.6 kJ/mol calculated by reaction kinetics experiment, far superior to that of Na-free counterpart (199 h−1 and 93.1 kJ/mol, respectively). The responsive experiments of water injection reveal that the water molecules are helpful for the oxidation reaction of BA and promote the conversion of BA. More importantly, the Na-modulated catalyst can further enhance the promotion effects of water in the reaction. The Na-modulated catalyst can obviously improve the dispersion of Pd nanoparticles. Also, the Pd active sites present a high oxidation state, and the Pd catalyst modulated by alkali cation can form abundant Pd−OHx species with high activity. The abundant Pd−OHx is helpful to enhance the water-induced reaction process. These finds can provide an avenue to enhance the chemical reactions involving water promotion.
To simplify the synthesis of the catalyst with alkali metals, the catalyst precursors with graphite oxide were prepared using NaOH as alkali source by the coprecipitation-calcination method (Scheme S1 in Supporting information). Correspondingly, the residual Na in the washing process can be controlled, and the Pd catalysts were synthesized via the facile wet impregnation method. X-ray diffraction (XRD) patterns and the FT-IR spectra (Fig. S1 in Supporting information) show that the precursors and catalysts can be assigned to the spinel structure of ZnCr2O4 and the Na atoms in Pd(Na)/rGO-ZnCr2O4 is stabilized by Na−O bonds. Meanwhile, the weak diffraction peaks of Pd are presented, indicating the uniform distribution of Pd with an ultrafine size on the surface of (Na)rGO-ZnCr2O4. The scanning electron microscope (SEM) images (Fig. S2 in Supporting information) indicate that the ZnCr2O4 grows uniformly on the GO surface by the nucleation and conversion of the pre-adsorbed Zn2+ and Cr3+ on GO after calcination, forming (Na)rGO-ZnCr2O4 containing 9% content of rGO, as confirmed by thermogravimetric analysis (TGA) results in Fig. S3 (Supporting information). In addition, the surface wettability of Pd(Na)/rGO-ZnCr2O4 exhibits a similar affinity for toluene and water, which favor the formation of stable Pickering emulsion (Fig. S4 in Supporting information).
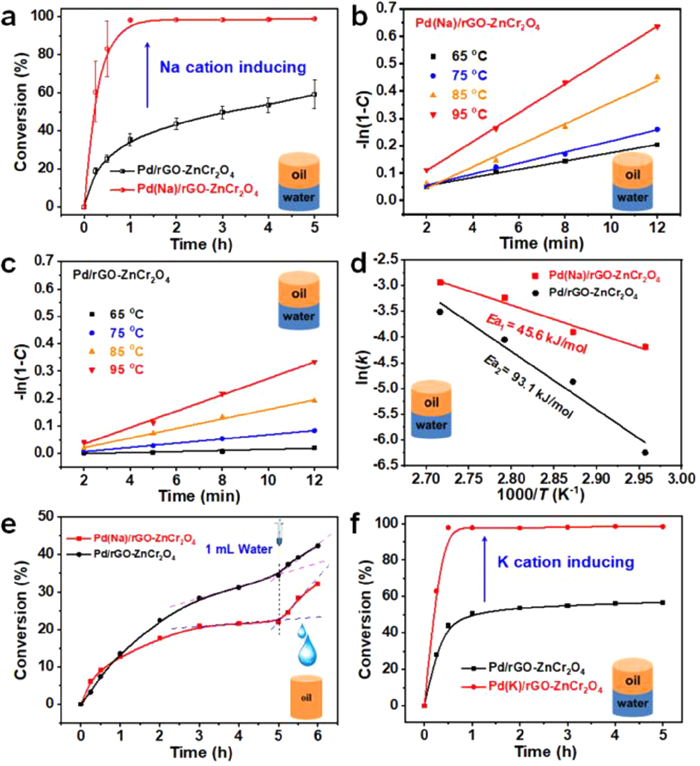
To compare the reactivity of Na-free and Na-modulated catalysts, the BA oxidation was chosen as a model reaction, based on our previous work [7,30]. As shown in Fig. 1a, compared with that of the Pd/rGO-ZnCr2O4 (a conversion of 36% and selectivity of ~99%), the Pd(Na)/rGO-ZnCr2O4 exhibits a superior catalytic activity, with a conversion of 98% and selectivity of ~99% at 1 h, being an increase in TOF from 199 h−1 to 469 h−1. The high selectivity for benzaldehyde is consistent with our previous work [7,31]. Furthermore, the macroscopic kinetic experiments of catalysts were performed and the conversion-time plots were obtained at different reaction temperatures (65, 75, 85, 95 ℃) and shown in Figs. 1b and c. The −ln(1 − C) of catalysts and reaction time present a linear relationship with R2 > > 0.99, which indicates that the oxidation of BA follows first-order kinetics. The rate constant (k) can be obtained from the slope and the corresponding k value is 0.039 min−1 for Pd(Na)/rGO-ZnCr2O4 and 0.017 min−1 for Pd/rGO-ZnCr2O4 at 85 ℃, respectively, which further indicates that the catalytic performance of Pd(Na)/rGO-ZnCr2O4 is superior of the Pd/rGO-ZnCr2O4 catalyst. The ln(k) and 1000/T at different temperatures also exhibit a linear relationship with R2 > 0.95, and the apparent activation energy (Ea) is collected (Fig. 1d). The Pd(Na)/rGO-ZnCr2O4 (45.6 kJ/mol) presents a low Ea value in comparison to that of Pd/rGO-ZnCr2O4 (93.1 kJ/mol), which indicates that the Na cations over Pd(Na)/rGO-ZnCr2O4 can promote and enhance the catalytic performance for BA.
Figure 1
Figure 1.
(a) Catalytic performance over Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4. (b, c) Time-conversion plots at various temperatures for Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4 catalyst, respectively. (d) Arrhenius plots for BA oxidation. (e) Response of water injection over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst for BA oxidation, with toluene as reaction solvent. (f) Catalytic activity test over Pd(K)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4.
Further, the Na-promoted effects for BA oxidation are well extended to other supports such as Pd(Na)/ZnCr2O4 Pd/ZnCr2O4 with rGO-free catalysts. The catalytic performance of Pd(Na)/ZnCr2O4 (Fig. S5a in Supporting information) is distinctly superior to Pd/ZnCr2O4 in the oxidation process of BA, and the corresponding macroscopic kinetic experiments were also performed (Figs. S5b–d in Supporting information). The Ea values of Pd(Na)/ZnCr2O4 and Pd/ZnCr2O4 catalysts are 66.6 and 129.7 kJ/mol, respectively. This further confirms that the Na cations over the catalyst have a certain positive effect, and contribute to the oxidation of BA. Moreover, the catalytic performance of the Pd catalysts using (Na)rGO-ZnCr2O4 as support is better than that of Pd(Na)/ZnCr2O4 (Figs. S5a and S6 in Supporting information), which suggests that the rGO has a certain promotion effect on the present reaction. The corresponding details for the synergistic effects were confirmed and demonstrated in Figs. S7–S9 (Supporting information). Furthermore, the TOF values over various catalysts were calculated, which presents a linear relationship (R2 = 0.99) with the molar ratio of Na to Pd (nNa: nPd) in the catalysts (Fig. S10 in Supporting information). That is to say, the amount of Na cations over the catalysts is inseparable from the activity of the catalyst.
In addition, numerous studies have also reported that water participates in catalytic reactions and can promote chemical reaction, such as CO oxidation [32–34], alcohol oxidation [7,35,36], and some hydrogenation reactions [37]. Furthermore, the water-promoted effects for BA oxidation catalyzed by Pd(Na)/rGO@ZnCr2O4 and Na-free counterpart catalyst were investigated. After the conversion rate of BA almost reaches a plateaus within 5 h only using toluene as a solvent, 1 mL water was injected, and the results are presented in Fig. 1e. The catalytic activities of Pd/rGO-ZnCr2O4 and Pd(Na)/rGO-ZnCr2O4 in the pure toluene system only has a reduction compared to that in Pickering emulsion within 5 h (Fig. 1a), which is more obvious for Pd(Na)/rGO-ZnCr2O4. When adding 1 mL water, the BA is further converted to benzaldehyde, which manifests that the water molecules promote the chemical reaction process. Also, the conversion rate of BA is faster over Pd(Na)/rGO-ZnCr2O4 than that on Pd/rGO-ZnCr2O4. That is to say, the water-promoted effects are more obvious and further promoted over catalyst with Na species, which is also one of the reasons why the catalytic activity of Pd(Na)/rGO-ZnCr2O4 is superior to that of Pd/rGO-ZnCr2O4 in Pickering emulsion reaction system. In general, the water in the system can contribute to the conversion of BA. The Na cation modified-catalyst can effectively further enhance the promotion effects of water molecules in the oxidation of BA. In addition, the Na cations in catalyst are also replaced by K cations, and the similar promotion effects are also observed (Fig. 1f), which further indicates that with the alkali cations in the catalyst, the reaction barrier of the rate-determining step is effectively reduced.
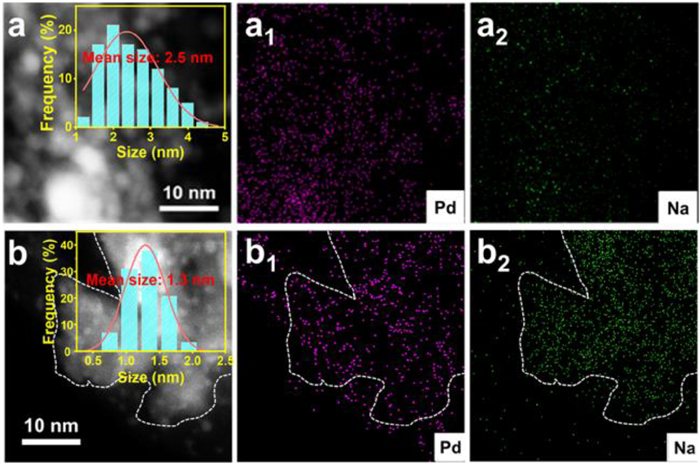
As we know, the particle size of active metal has some effects on the activity of catalysts [38,39]. The transmission electron microscopy (TEM) was performed to detect the morphology and size distribution of Pd nanoparticles on Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4, and the results were shown in Fig. S11 (Supporting information). It was found that the Pd nanoparticles with ultrafine size are evenly dispersed on the surface of the support without the distinct lattice fringe, which were further analyzed by high-angle annular dark-field scanning TEM (HAADF-TEM). The Pd average size for Pd(Na)/rGO-ZnCr2O4 and Pd(Na)/rGO-ZnCr2O4 is statistically 1.3 nm and 2.5 nm, respectively, as shown in Fig. 2 and Fig. S12 (Supporting information). It is indicated that the Na cation can induce the dispersion of Pd nanoparticles on the surface of Pd(Na)/rGO-ZnCr2O4. In fact, the analogous behavior was also found and confirmed in the past [13]. The corresponding elemental mapping images also reveal that the produced catalyst presents the uniform distribution of Pd and Na over the surface of catalyst. To better present the promotion effects of Na for Pd dispersion, CO pulse experiments were conducted to reveal the dispersion of metal, the ratio of active metal atoms to total metal atoms on the catalyst (Figs. S13a and b in Supporting information). The metal dispersion of Pd(Na)/rGO-ZnCr2O4 (54%) is superior to that of Pd/rGO-ZnCr2O4 (39%) and similar results can be found for the corresponding rGO-free catalyst, which further confirms that the alkali cations in catalyst can promote the dispersion of Pd atoms. In addition, the metal dispersions of Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4 are superior to that of the corresponding rGO-free counterpart catalysts (Figs. S13c and d in Supporting information), respectively, maybe resulting from the synergistic interaction rGO and ZnCr2O4. The above results disclose that the Na cations in the catalyst indeed enable the dispersion of Pd active sites, which can expose the abundant active sites to enhance the oxidation of BA.
Figure 2
Figure 2.
HAADF-STEM images of (a) Pd/rGO-ZnCr2O4 and (b) Pd(Na)/rGO-ZnCr2O4 and the corresponding curves of Pd size distribution (inset). (a1, b1 and a2, b2) EDS element maps of Pd and Na for Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4, respectively.
Additionally, the O 1s region for X-ray photoelectron spectroscopy (XPS) spectra for all catalysts (Fig. 3a and Fig. S14 in Supporting information) reveals three types of oxygen species (denoted as O1, O2, and O3), which can be attributed to lattice O* (529.0–530.0 eV), lattice OH bonded with metal atoms (530.5–531.5 eV) and crystalline H2O (532.1–533.0 eV) [22,40], respectively. Compared with Pd/rGO-ZnCr2O4, the O1 and O3 peaks of Pd(Na)/rGO-ZnCr2O4 have a shift to low and high binding energy, respectively. The electronic properties of the Pd(Na)/rGO-ZnCr2O4 surface can be altered. In addition, the O2 region area for Pd(Na)/ZnCr2O4 and Pd(Na)/rGO-ZnCr2O4 catalysts is larger than that of the corresponding Na-free catalyst. This suggests that the appearance of Na cations significantly increases the density of lattice OH, forming the active Pd−OHx structure analogously described in the literature [11,12]. The abundant surface OH can also serve as the adsorption sites for BA to contribute to the chemical oxidation reaction [41]. In addition, the Na KLL Auger peak signal can be seen at the 534–536 eV toward the Pd(Na)/rGO-ZnCr2O4 and Pd(Na)/ZnCr2O4 catalysts (Fig. 3a and Fig. S14). The Na 1s XPS spectra (Fig. 3b) reveal that there is only one major peak centered at 1071.6 eV, implying that the sodium species bind to the surrounding substrate through –O–Na linkages [14], which is consistent with the results of FTIR spectra (Fig. S1b in Supporting information). Nevertheless, the relatively weak Na 1s signal for Pd/rGO-ZnCr2O4 is presented, which derives from a small amount of remained Na species within the catalyst. The ICP results (Table S1 in Supporting information) further reveal that the content of Na in Pd/rGO-ZnCr2O4 and Pd(Na)/rGO-ZnCr2O4 are 0.9 and 3.3 wt%, respectively.
Figure 3
Figure 3.
The XPS spectra of (a) O 1s, (b) Na 1s and (c) Pd 3d for Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst. (d) In situ DRIFTS of CO chemisorption over Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst.
To eliminate the effects of residual Na in the catalyst, the precursor of catalyst was also synthesized by NH3·H2O as a coprecipitation reagent instead of NaOH, and meanwhile the catalysts with the different molar ratios of Na to Pd were also prepared. The corresponding catalytic performances were shown in Fig. S15 (Supporting information). It is observed that the activity gradually improves with an increase of Na content in the catalyst, indicating that the performance of catalyst is closely correlated with the content of Na species in the catalyst. The TOF values for the various catalysts were calculated, showing a linear relationship (R2 = 0.92) with the molar ratio of Na to Pd within the catalysts. Furthermore, the XPS spectra (Fig. S16 in Supporting information) reveal that the O1 and O3 peaks of the catalysts with an increase of Na content have a shift to low and high binding energy, respectively. The O2 (a symbol of Pd−OHx structure) region area gradually increases with an increase of Na content in the catalyst, reaching the peak value at the molar ratio of Na to Pd of 5:1 and keeps constant. The above results further reveal that the Na cations in the catalyst can change the surface electron properties of the catalyst, creating the highly active Pd−OHx species. The activities of catalysts are enhanced with increased molar ratio of Na to Pd and almost reaches the maximum at 5:1. Further, at 7:1, the activity only has a slight change. The corresponding changes are consistent with the O2 region area of the catalysts. This further confirms that the alkali-stabilized Pd−OHx species contribute to enhancing the activity of catalyst. The increased content of Na in the catalyst can be also supported by the Na 1s XPS spectra (Fig. S16b).
It has also been reported that the alkali, as catalyst promoters, can affect the oxidation state of active metals [9,10]. Furthermore, the high-resolution spectra of the Pd 3d region for Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4 (Fig. 3c) can be deconvoluted with binding energies of 335.9, 337.4 eV and 341.2, 342.7 eV, which correspond to Pd0 and Pd2+, respectively. The fraction of Pd2+/Pd0 for Pd(Na)/rGO-ZnCr2O4 (1.52) on the basis of the peak area has a dramatic increase in comparison to that of Pd/rGO-ZnCr2O4 (0.76), which indicated that the Na cation in catalysts can tune the electronic structure of (Na)rGO-ZnCr2O4 surface and the Pd species exist in the oxidation states resulting from the effects of Pd−OHx species. This can be confirmed by the increased fraction of Pd2+/Pd0 with the increase of Na content in the prepared catalysts using NH3·H2O with different molar ratios of Na to Pd (Fig. S17 in Supporting information), where the peak corresponding to Pd2+ shows a shift toward a higher binding energy than that of Na-free catalyst.
To further confirm the effects of Na cations on Pd active sites, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of CO chemisorption was performed. The four peaks are exhibited for Pd/rGO-ZnCr2O4, as presented in Fig. 3d. The bands at 2079 and 2050 cm−1 are assigned to linear adsorption of CO on catalyst [42]. The band at 1967 cm−1 is μ2-bridge adsorption of CO on the step and facet edges. The broad band around 1760–1944 cm−1 is considered as μ2-bridge adsorption of CO on (111) facet of Pd nanoparticles [43]. In the case of Pd(Na)/rGO-ZnCr2O4 with Na species, the absorption peaks shift toward low wavenumber, changing from 2079 cm−1 to 2073 cm−1, 1967 cm−1 to 1957 cm−1, and 1760–1944 cm−1 to 1722-1944 cm−1, indicative of the back-donation of Pd d-electrons to the 2π* antibonding orbital of CO [44]. The promotion of Pd electron-donation ability may result from the electronic effects of Pd−OHx structure triggered by Na cation. Furthermore, the positive effects of Na cations on Pd active sites are well extended to other catalysts such as Pd(Na)/ZnCr2O4 and Pd/ZnCr2O4, and the results were exhibited in Fig. S18 (Supporting information). Only the wide peak for μ2-bridge-bonded CO on (111) facets of Pd nanoparticles appears and shifts from 1757–1944 cm−1 to 1724–1944 cm−1. The reasons may be resulted from the low content of Na for Pd(Na)/ZnCr2O4 and Pd/ZnCr2O4 compared with Pd(Na)/rGO-ZnCr2O4 (Table S1). Overall, the Na cations in catalyst mainly modulate the electronic configuration of Pd active sites and further form the Pd−OHx on the surface of catalyst. Therefore, the Pd−OHx in Pd(Na)/rGO-ZnCr2O4 is closely related to and responsible for the enhanced catalytic activity.
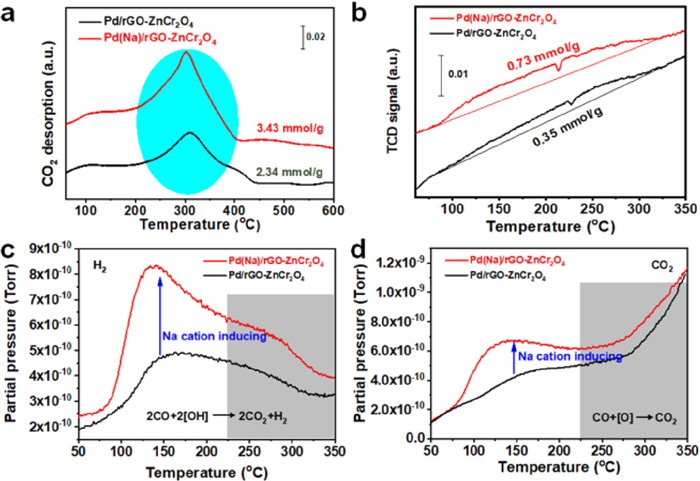
The concentration of base sites on the catalyst surface is also a significant factor to affect the oxidation of BA [45], in which benzyl alcohol can be adsorbed, and converted into benzaldehyde. Therefore, it is necessary to investigate the effects of Na cations on the base sites of the catalyst surface. As a result, CO2-TPD was performed and the results are shown in Fig. 4a and Fig. S9 (Supporting information). For CO2-TPD curves of supports, the wide peak at a low temperature of 60–200 ℃ can be found, which is due to the weak base sites of rGO-ZnCr2O4 or (Na)rGO-ZnCr2O4 and the weak physical interaction between CO2 and supports. Also, a weak and similar CO2 desorption signal corresponding to the strong base sites can also be found at high temperature of 200–450 ℃. This implies that the base sites on the surface of supports are unaffected by alkali cations. After anchoring Pd on (Na)rGO-ZnCr2O4 and rGO-ZnCr2O4, the peak at a low temperature still appears and strong peak of CO2 desorption corresponding to the strong base sites is observed at high temperature region. It can be inferred that the strong base sites are mainly attributed to the supported Pd nanoparticles. In addition, the total concentration of base sites for Pd(Na)/rGO-ZnCr2O4 (3.43 mmol/g) has a remarkable increase in comparison to that of Pd/rGO-ZnCr2O4 (2.34 mmol/g). The increased concentration of strong base sites is mainly attributed to the facts that the Na cations substantially enhance the dispersion of Pd nanoparticles and meanwhile facilitate the formation of the surface Pd−OHx series. The strong base sites on the Pd(Na)/rGO-ZnCr2O4 surface are beneficial to the realization of excellent catalytic performance for BA oxidation.
Figure 4
Figure 4.
(a) CO2 temperature-programmed desorption (CO2-TPD) profiles on Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst. (b) CO-TPR profiles of Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst, which corresponds to MS signal: (c) H2 and (d) CO2.
In the process of BA oxidation, the −OH on the catalyst surface can be helpful for the dissociation −O−H bonds of the BA and decrease the energy barrier of α-C−H bonds of the ensuing alkoxide intermediate to achieve the conversion toward benzaldehyde [41]. In the past, it has been reported that the introduction of alkali cation in Au and Pt catalysts can induce the production of surface hydroxyl groups [12,13]. To further confirm the presence of surface hydroxyl groups, the CO temperature-programmed reduction (TPR) coupled with mass spectrometry to track the off-gas, an effective way of identifying the surface −OH and active lattice [O] over catalysts (2CO + 2[OH] → 2CO2 + H2, CO + [O] → CO2), was performed and the results were exhibited in Fig. 4b. Compared to the Pd/rGO-ZnCr2O4, the Pd(Na)/rGO-ZnCr2O4 catalyst exhibits a similar TCD signal peak region at 85–310 ℃. However, the Na-containing catalyst has a significant increase toward CO consumption from 3.54 mmol/g to 7.26 mmol/g. Meanwhile, the CO-TPR experiments over the Pd/rGO-ZnCr2O4 and Pd(Na)/rGO-ZnCr2O4 catalysts were also carried out and the off-gas was traced by mass spectrometry, of which the results were presented in Figs. 4c and d. At a relatively low temperature (< 225 ℃), the produced H2 and CO2 molecules are mainly attributed to the reaction between CO and −OH species of catalyst surface. At high temperature of 225–350 ℃, it can also be found that the partial pressure of H2 decreases while that of CO2 increases. In this process, the decrease of H2 partial pressure results from the gradual consumption of −OH over the catalyst surface with an increase of temperature. The increased CO2 partial pressure comes from the reaction between CO and the active lattice [O] species on the catalysts surface. This indicates that the surface −OH over the catalysts can feature a higher activity than surface lattice [O] species at low temperature. It can be easily concluded that surface −OH has a stronger activity than surface reactive oxygen species, which manifests that the oxidation reaction of BA mainly follows the Langmuir-Hinshelwood mechanism rather than the Mars-van Krevelen mechanism involved in the active lattice [O] species. In addition, the Na-modified Pd(Na)/rGO-ZnCr2O4 shows a high partial pressure of H2 and CO2, and the peak position shifts to low temperature in comparison to Pd/rGO-ZnCr2O4 catalyst. This further indicates that the Na cations in catalyst benefit to provide the abundant −OH species. Further, the surface −OH species over Pd(Na)/rGO-ZnCr2O4 with high activity will easily bind the Pd species.
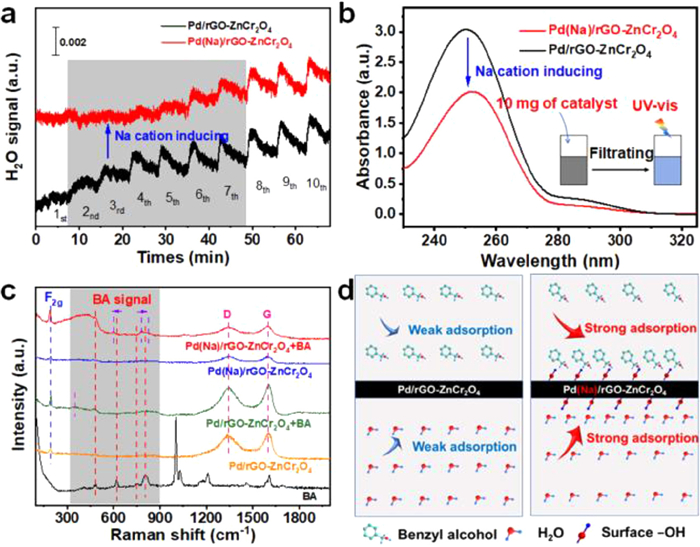
In addition, according to the results of water injection experiment, the water promotes the oxidation reaction of BA. Thus, it is necessary to investigate the adsorption of catalysts toward water. To decipher the relationship between H2O molecule and the Na cation modified Pd(Na)/rGO-ZnCr2O4, H2O pulse adsorption experiments were carried out over Pd(Na)/rGO-ZnCr2O4 and the counterpart catalyst, and the results were presented in Fig. 5a. The Pd/rGO-ZnCr2O4 almost reaches adsorption saturation for H2O after suffering from H2O vapor pulse for three times. Comparably, the Na-modified Pd(Na)/rGO-ZnCr2O4 catalyst undergoes H2O vapor pulse for seven times and can reach adsorption saturation. This suggests the enhanced adsorption of H2O molecules in the presence of Na cations, which maybe result from the hydrogen bond interaction between H2O molecules and the new appearing surface Pd−OHx species. Further, the adsorption characteristics of BA on catalyst surface were investigated by UV-vis absorption experiments. The BA in the filtrate adsorbed by Na-modified Pd(Na)/rGO-ZnCr2O4 shows a lower absorbance than that of the Pd/rGO-ZnCr2O4 (Fig. 5b), which shows that the Na cations in catalyst can improve the absorption capacity of catalyst toward BA. To gain the detailed adsorption information of BA over catalyst surface, in situ Raman experiments were performed (Fig. 5c). For Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4 without BA, the similar peaks at 193 (δ(Zn−O)), 1344 (D bond), and 1600 cm−1 (G band) are observed. When BA is adsorbed on the Pd(Na)/rGO-ZnCr2O4, it can be found that there are four characteristic peaks of BA around 400 cm−1 to 900 cm−1. The peak at 485 cm−1 corresponding to out-of-plane deformation of the phenyl ring for BA is almost unchanged. While the peak at 620 cm−1, being in-plane bending vibration of the phenyl ring, shifts to low wavenumber of 601 cm−1. Also, the −C−H bending vibration at the phenyl ring shifts from 750 cm−1 to 784 cm−1 and 807 cm−1 to 827 cm−1, respectively. The shifted peaks are from the distorted/twisted BA that is adsorbed on the catalyst, thus the corresponding reactive barrier of BA to benzaldehyde is reduced. For Pd/rGO-ZnCr2O4 catalyst with BA, the peak at 485 cm−1 belonging to out-of-plane distorted phenyl ring can be recognized. The other peaks assigned to BA molecules are relatively weak in comparison to that of Pd(Na)/rGO-ZnCr2O4. It is easy to conclude that the adsorption of Pd/rGO-ZnCr2O4 for BA is relatively weak than that of Pd(Na)/rGO-ZnCr2O4 modified by the Na species. In other words, the Na-modified Pd(Na)/rGO-ZnCr2O4 catalyst can enhance the adsorption of H2O molecule on account of the Pd−OHx structure induced by Na cations compared to the corresponding Na-free sample. Also, the changed adsorption due to the distorted structure of BA and improved adsorption capacity for BA on the catalyst with Na species are synergistically beneficial for the conversion of BA. This is also visualized and shown in Fig. 5d.
Figure 5
Figure 5.
(a) H2O pulse curves over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free sample. (b) UV-vis absorption spectra for aqueous solutions after BA is adsorbed over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free sample. (c) Raman spectra of BA, the various catalysts (Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4) after adsorbing BA. (d) Adsorption kinetics models of BA and H2O over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free sample.
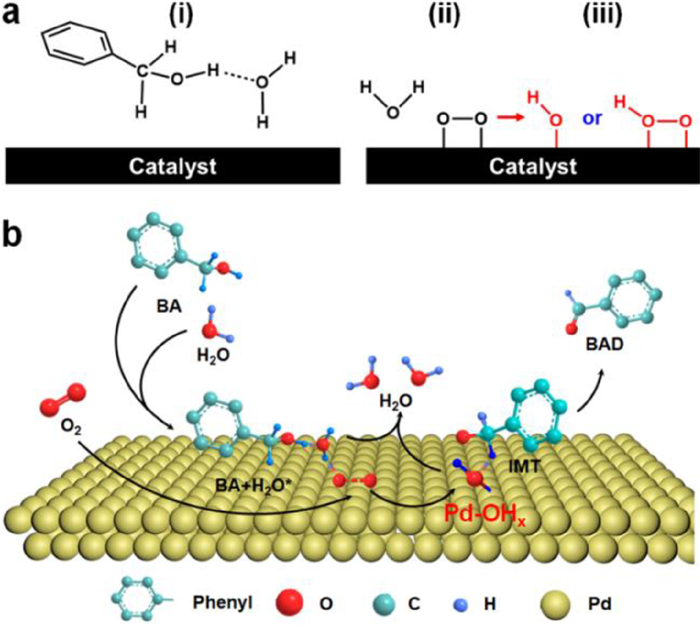
For the oxidation of BA to benzaldehyde, the reaction process involves the cleavage of α−C−H and −O−H bonds of BA. It was confirmed that H2O molecules in the reaction system feature the multiple effects during BA oxidation as follows (Fig. 6a): (ⅰ) Enhancing adsorption towards BA derived from being distorted and twisted BA on the surface of catalyst [7], (ⅱ) enhancing adsorption and activation of O2 [46], (ⅲ) generating active *OH or *OOH formed by the reaction of H2O and O2, where the *OH or *OOH can further react with the hydrogen of α−C−H [36,41]. Also, when the catalyst is modified with a reasonable amount of alkali cations, the surface of the catalyst can feature the abundant high active Pd−OHx species compared with Na-free catalyst, which has been confirmed by the previous experiment. Further, the catalyst with abundant high active Pd−OHx exhibits more excellent catalytic performance for BA oxidation than Na-free catalyst under the same reaction conditions. It can be imagined that the −OH in alkali-stabilized Pd−OHx would function as *OH or *OOH to some degree. In this case, the reaction can be further promoted, as discussed in the results. Based on this, the corresponding mechanism of alkali-promoted alcohol oxidation can be proposed following the Langmuir-Hinshelwood mechanism and the detailed reaction is schemed in Fig. 6b. The BA and H2O linked by hydrogen bond interaction (*H2O···HO−CH2−C6H5) are easily adsorbed on the surface of the catalyst [7]. The H3O* formed by the −O−H cleavage of *H2O···HO−CH2−C6H5 on active sites of catalyst surface combines with the activated oxygen decomposed by O2 on the catalyst surface to form the H2O molecule, *OH or *OOH species, and intermediate (IMT) C6H5−CH2O* [32,36]. While the hydrogen in the α−C−H of IMT would efficiently combine with the abundant high active Pd−OHx or *OH and *OOH to form C6H5−CHO and H2O. Moreover, it was reported that the consumed −OH for Pd−OHx catalyst in the reaction can be supplemented via*OOH or *OH species derived from the reaction of H3O* and the activated oxygen decomposed by O2. The roles of alkali in the catalyst are mainly to provide the abundant high active Pd−OHx, resulting in excellent catalytic performance.
Figure 6
Figure 6.
(a) Schematic diagram for water-induced oxidation of BA. (b) Scheme of the detailed reaction for BA oxidation on the surface of abundant Pd−OHx. For a better view, only one −OH on the Pd surface is drawn in the schematic.
In conclusion, the positive effects of alkali cations on Pd catalyst for oxidation of BA are investigated and deciphered in this work. The macroscopic kinetic experiments confirm that rational Na-containing catalyst can decrease the apparent activation energy of BA oxidation reaction from 93.1 kJ/mol to 46.3 kJ/mol resulting from the enhanced water-induced reaction process by the Na-modified catalyst. It is well recognized and identified that the Na cations over the catalyst can decrease the size of Pd nanoparticles and promote the dispersity of Pd active sites. The Na species are present over the catalyst in the form of Na−O linkage. Also, the Pd active sites show a high oxidation state compared with that of Na-free catalyst, and the surface of Na-modified catalyst presents the abundant −OH species by the formation of Pd−OHx. This is helpful for the adsorption of water and BA, further leading to the distorted BA with high reaction activity. The abundant highly active Pd−OHx species derived from the effects of Na cations can efficiently combine with the hydrogen of α−C−H for C6H5−CH2O* intermediate, which is responsible for excellent catalytic performance for BA oxidation. It is expected that the strategy of alkali cations-modified catalysts would provide an avenue to promote and enhance the chemical reactions involved in water-induced effects including CO oxidation, alcohol oxidation, hydrogenation reaction, etc.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC, Nos. 51872035 and 22078052) and the Innovation Program of Dalian City of Liaoning Province (No. 2019RJ03).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107939.
[1]
S. Crossley, J. Faria, M. Shen, D.E. Resasco, Science 327 (2010) 68–72. doi: 10.1126/science.1180769
[2]
D. Tian, X. Zhang, H. Shi, et al., J. Am. Chem. Soc. 143 (2021) 16641–16652. doi: 10.1021/jacs.1c07482
[3]
X. Zhang, Y. Hou, R. Ettelaie, et al., J. Am. Chem. Soc. 141 (2019) 5220–5230. doi: 10.1021/jacs.8b11860
J. Saavedra, T. Whittaker, Z. Chen, et al., Nat. Chem. 8 (2016) 584–589. doi: 10.1038/nchem.2494
Figure 1
(a) Catalytic performance over Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4. (b, c) Time-conversion plots at various temperatures for Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4 catalyst, respectively. (d) Arrhenius plots for BA oxidation. (e) Response of water injection over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst for BA oxidation, with toluene as reaction solvent. (f) Catalytic activity test over Pd(K)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4.
Figure 2
HAADF-STEM images of (a) Pd/rGO-ZnCr2O4 and (b) Pd(Na)/rGO-ZnCr2O4 and the corresponding curves of Pd size distribution (inset). (a1, b1 and a2, b2) EDS element maps of Pd and Na for Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4, respectively.
Figure 3
The XPS spectra of (a) O 1s, (b) Na 1s and (c) Pd 3d for Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst. (d) In situ DRIFTS of CO chemisorption over Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst.
Figure 4
(a) CO2 temperature-programmed desorption (CO2-TPD) profiles on Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst. (b) CO-TPR profiles of Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free catalyst, which corresponds to MS signal: (c) H2 and (d) CO2.
Figure 5
(a) H2O pulse curves over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free sample. (b) UV-vis absorption spectra for aqueous solutions after BA is adsorbed over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free sample. (c) Raman spectra of BA, the various catalysts (Pd(Na)/rGO-ZnCr2O4 and Pd/rGO-ZnCr2O4) after adsorbing BA. (d) Adsorption kinetics models of BA and H2O over the Pd(Na)/rGO-ZnCr2O4 and the corresponding Na-free sample.
Figure 6
(a) Schematic diagram for water-induced oxidation of BA. (b) Scheme of the detailed reaction for BA oxidation on the surface of abundant Pd−OHx. For a better view, only one −OH on the Pd surface is drawn in the schematic.

 DownLoad:
DownLoad:


 下载:
下载:







