CAS Key Laboratory of Design and Assembly of Functional Nanostructures, and Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Xiamen 361021, China
b.
Xiamen Institute of Rare Earth Materials, Haixi Institute, Chinese Academy of Sciences, Xiamen 361021, China
c.
Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou 350108, China
Received Date:
06 June 2022 Accepted Date:
21 June 2022 Revised Date:
16 June 2022 Available Online:
15 June 2023
Abstract:
Developing redox switches that not only perform specific mechanical movements but also drive important chemical reactions is important but a great challenge. Herein, we report a redox pair of cobalt species (CoⅢ/CoⅡ) that switches through photo-dehydrogenation of alcohol and hydrogenation of azo-ligand. The cobalt species is equipped with a flexible azo-ligand containing two bulky planar substituents. A planar oxidated sate (CoⅢ species) can be photo-reduced to a saddle-like reduced state (CoⅡ) with alcohol molecules as electron donors, and in turn the CoⅢ species can be recovered with azo-ligand as oxidant under acidic surrounding. Both the redox states of the pair are isolated and characterized by single crystal X-ray diffraction. In the switching cycle, alcohol is oxidized to aldehyde by azo-ligand through proton coupled electron transfer and the cobalt complex acts as a redox catalyst. These results provide important insights into alcohol dehydrogenation catalyzed by redox complexes.
Mechanical molecular switches, manifesting reversible conformational changes induced by a particular stimulus, have received a great attention for their fascinating functionalities [1-4]. A variety of external stimuli, including thermal [5-7], light-irradiation [4,7-9], pH [10,11], and coordination of metal ions [12] have been applied to induce molecular conformational changes. Reversible molecular conformation changes induced by redox produce redox switches [2,13-15]. A lot of intriguing conformational variations have been achieved by controlling the oxidation states of corresponding constitution, resulting in the course of movements in rotaxanes [15], rotary motion [16], helix inversion [17], and intramolecular anion translocation [18].
More importantly, redox switches serve as electron carriers to transport electrons between different reactants. Thus, they also drive redox reactions during their own conformational changes [19,20]. Coordination compounds with redox active metal ions coordinated by flexible ligands are good candidates for redox switches. The variation of oxidation states of metal ions would stimulate the mechanical movement of a flexible ligand. In turn, the redox potential of the metal ion can be finely adjusted by the conformational variation of the ligands. Therefore, some certain reactions can be driven by the redox switching cycle. For instance, Chen et al. demonstrated a pair of diiridium bimetallic complexes acting as redox switch to directly split carbonate to carbon monoxide and oxygen through a low energy pathway [20]. However, the rational design of a redox switch that drives a certain reaction remains a great challenge.
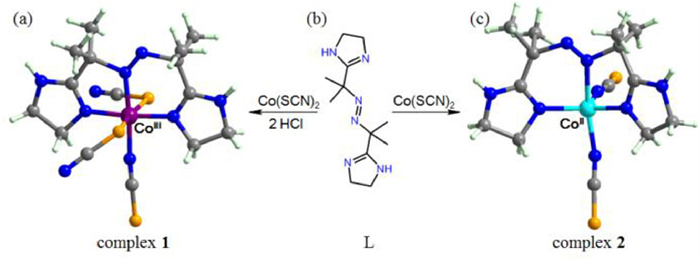
In this study, we report a redox pair of cobalt species LCoⅢ(SCN)3 (1) and LCoⅡ(SCN)2 (2) (L = 2,2′-azobis[2-(imidazolin-2-yl)propane) exhibiting redox coupled mechanical motion. The intrinsic flexibility of L might make it capable of adapting the coordination of both the CoⅢ and CoⅡ ions, therefore both the states of the redox pair are isolated and characterized by single crystal X-ray diffraction. The redox pair switches through photo-dehydrogenation of alcohol and hydrogenation of azo-ligand. It should be noted that alcohol dehydrogenation is an important reaction in the bacterial alcohol metabolism cycle [21-24] and a variety of processes for manufacturing chemicals, pharmaceuticals, foods, and fuels [25-32].
Single crystals of a (CoⅢ/CoⅡ) redox couple LCoⅢ(SCN)3·0.2H2O (1) and LCoⅡ(SCN)2 (2) were synthesized by the ambient reaction of Co(SCN)2 with L·2HCl and L, respectively (Fig. 1). Complex 1 also can be obtained by the same reaction under an inert condition. The acidic environment is critical for the formation of complex 1, implying proton is critical for the oxidation of the original CoⅡ ions. Oxidizing complex 2 with di–tert–butyl azodicarboxylate (DBAD) under acidic surrounding leads to complex 1 and the corresponding hydrazine. The produced hydrazine has been isolated and characterized by single crystallography (Fig. S1 in Supporting information). This result indicates the CoⅡ ions are probably oxidized by the L ligand via a process of proton coupled electron transfer (PCET) during the synthesis of complex 1 [33,34]. The Infra-red (IR) spectra, powder X-ray diffraction (PXRD) patterns and thermogravimetric analyses (TGA) for the CoⅢ/CoⅡ redox couple are presented in Figs. S2–S4 (Supporting information).
Figure 1
Figure 1.
Synthesis scheme and crystal structures of complexes 1 and 2. (a) Crystal structure of complex 1. (b) Chemical structure of azo ligand L. (c) Crystal structure of complex 2. (C: gray, N: blue, S: orange, H: light green, CoⅢ: violet, CoⅡ: turquoise).
Complex 1 crystallizes in the mononuclear space group C2/c (Table S1 in Supporting information), with the two imidazolin rings from the L ligand almost coplanar with a dihedral angel of 178.4°. The CoⅢ ion is coordinated by three N atoms (one azo N atom and two imidazolin N atoms) from the L ligand, one N atom and two S atoms from three different SCN− ions (Fig. 1a) in a distorted octahedral geometry. The average Co−N and Co−S bond lengths are 1.901 Å and 2.318 Å, respectively, close to those of the reported low-spin CoⅢ complexes [35]. The low-spin of the CoⅢ in complex 1 is confirmed by the χmT value close to zero (Fig. S5 in Supporting information). Complex 2 crystallizes in the orthorhombic space group Cmca. Different from the planar L ligand in complex 1, the L ligand in complex 2 is saddle-like with the dihedral angel between the two imidazolin rings of 133.4°. The high-spin CoⅡ ion (χmT = 2.26 cm3 K/mol at 300 K) in complex 2 is coordinated by five N atoms (three from the L ligand and the other two from two different SCN− ions) (Fig. 1b) in a trigonal-bipyramidal coordination geometry. The Co−N bond lengths are in the range of 1.992−2.214 Å, comparable to those found in other high-spin CoⅡ complexes [36].
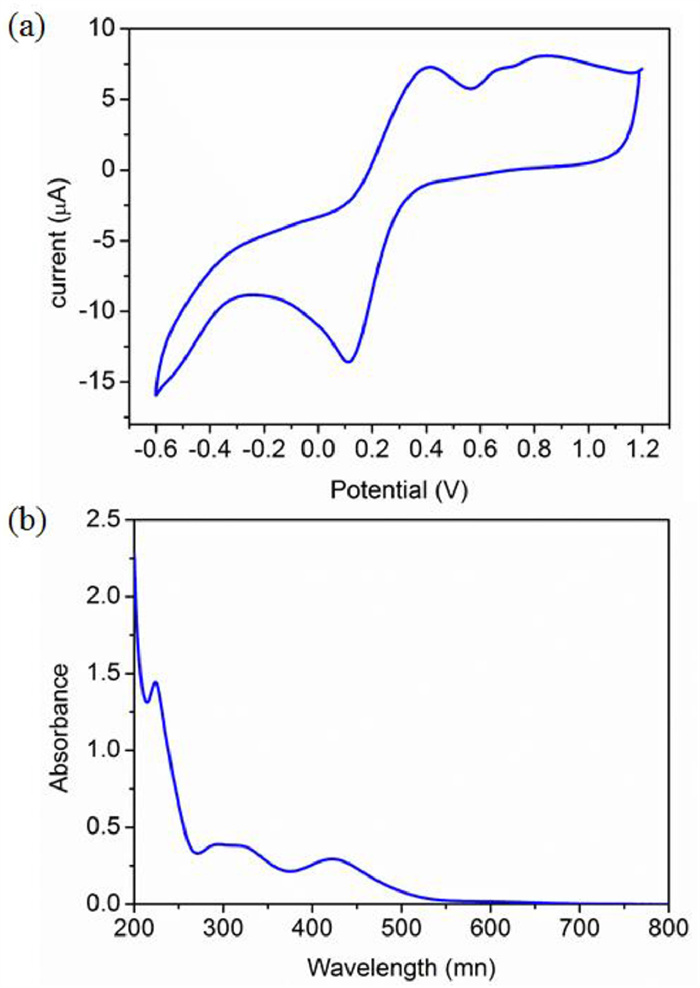
The stability of both the redox states enables it to be potential for the application as a chemical-driven molecular switch. The Electrospray Ionization Mass Spectrometry (ESI-MS) of complex 1 in CH3CN indicates [LCoⅢ(SCN)]2+ is the major CoⅢ species in the solution (Fig. S6 in Supporting information). The cyclic voltammetry (CV) of the above solution shows an obvious catalytic current with E1/2 = 0.26 V (Fig. 2a), which attributes to the redox process between the [LCoⅢ(SCN)]2+ and [LCoⅡ(SCN)]+ species. The UV–visible (UV–vis) absorption spectrum (Fig. 2b) of complex 1 in CH3CN displays a strong absorption band at 222 nm assigned to the intraligand π–π* transition, and two broad absorption bands at 306 and 419 nm corresponding to the ligand-to-metal charge transfer (LMCT). The strong band at 222 nm is also obvious in the spectra of the ligand L and complex 2 (Fig. S7 in Supporting information). Based on the optical and electrochemical observations, we envisioned that [LCoⅢ(SCN)]2+ can be facilely photoexcited and then reduced to [LCoⅡ(SCN)]+ if an appropriate electron donor accessible.
Figure 2
Figure 2.
(a) Cyclic voltammogram of 0.5 mmol/L complex 1 in acetonitrile and (b) absorption spectra of 0.5 mmol/L complex 1 in acetonitrile.
The cobalt redox couple is reminiscent the transitional metal catalysts for alcohol dehydrogenation based on pincer ligands [37-39]. A signal assigned to {[LCo(SCN)]MeOH}+ can be clearly observed by the HR-ESI-MS (Fig. S8 in Supporting information) measurement performed on complex 1 in MeOH implying MeOH is a promising reductant. Therefore, MeOH was initially selected as electron donor to reduce [LCoⅢ(SCN)]2+ to [LCoⅡ(SCN)]+. The photoreaction of complex 1 (5 mg, 0.01 mmol) and MeOH (500 equivalents) was firstly performed in CH3CN with one equivalent of tetrabutylammonium hydroxide to accept the generated protons during the reaction. Pink crystals of complex 2 were obtained with a yield ~72.5%. Remarkably, alcohol, alkali, and light irradiation are all necessary to the redox switching. The poor solubility of complex 2 in CH3CN prohibited us from in-situ studying the reversibility of the redox switching. Acetone attracted our attention because of its lower polarity and ability to accept proton. ESI-MS measurement (Fig. S9 in Supporting information) indicates the major CoⅢ species of complex 1 in acetone is also [LCo(SCN)]2+. In contrast to the CH3CN solution, the switching from [LCoⅢ(SCN)]2+ to [LCoⅡ(SCN)]+ can proceed in the absence of external alkali. Upon irradiation, the acetone solution of the reactant mixture (5 mg complex 1 and 500 equiv. MeOH) gradually changes from brown to blue without precipitate appearing. ESI-MS spectrum (Fig. S10 in Supporting information) shows clear signal (Exp. m/z = 367.1, Calcd. m/z = 367.1) of [LCo(SCN)]+, and crystals of complex 2 can be harvested with a yield ~55.3% upon crystallization by solvent evaporation in air. The reverse switching can be induced by azo-ligand slowly which acts as H atom acceptor. Therefore, a couple of H atoms transfer from alcohol to azo-ligand generating the corresponding aldehyde and hydrazine in the switching cycle.
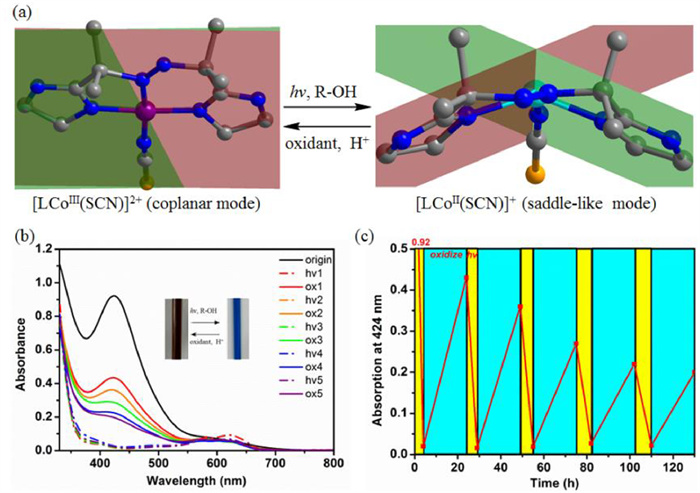
Then, we use UV–vis spectrum to monitor the switching between [LCoⅢ(SCN)]2+ and [LCoⅡ(SCN)]+ in acetone with cyclohexanol as the reductant and DBAD as the oxidant (Fig. 3). A solution of complex 1 (5 mmol/L) and cyclohexanol (500 equiv.) in 1.6 mL acetone was prepared, 0.1 mL of which was diluted in 3 mL acetone for UV–vis absorption measurement. The spectrum of the starting brown solution characteristic of a strong band at 424 nm remains unchanged when the solution is kept in dark (Fig. S11 in Supporting information). After irradiation for 4 h, the brown solution changes to blue, and the band at 424 nm almost disappears with a new weak band advent at 622 nm. The band at 424 nm reappears but reaches only half of the original intensity after adding one equivalent of DBAD. This result indicates only half of the complex can be recycled after a switching cycle. It is worth to note that the band at 424 nm spontaneously declines under irradiation. These results indicate the cobalt species partially decompose under irradiation. After ten switching cycles, the band at 424 nm recovers to ~12% of the original intensity (Fig. S12 in Supporting information). The amount of produced cyclohexanone after ten switching cycles was determined by gas chromatography-mass (GCMS) analysis (Fig. S13 in Supporting information). The result revealed ~0.1 mmol cyclohexanol is produced by 0.01 mmol cobalt species, corresponding to at least ten switching cycles of each cobalt species. Massive excessive DBAD prohibits the start of the redox switching, implying excessive azo group may occupy the active sites of [LCoⅢ(SCN)]2+ to prohibit alcohol to coordinate with the CoⅢ ion.
Figure 3
Figure 3.
Redox reaction driven mechanical motion of the Co switch. (a) Interconversion between two conformations of [LCoⅢ(SCN)]2+ (coplanar mode) and [LCoⅡ(SCN)]+ (saddle-like mode). (b) UV–vis absorption for photo-reduction (hv) of [LCoⅢ(SCN)]2+ and DBAD oxidation (ox) of [LCoⅡ(SCN)]+. (c) Reversible conversional cycles between the CoⅡ and CoⅢ species.
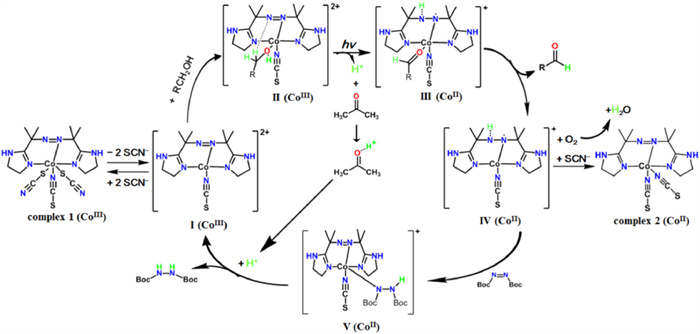
Based on the above observations, we deduced that the redox switching driven by alcohol oxidation and DBAD reduction is a metal-ligand cooperative mediated process [40], and the plausible mechanism is outlined in Scheme 1. Initially, two SCN− anions on the axial positions escape from complex 1 leading to the active species [LCoⅢ(SCN)]2+ (Ⅰ), which binds to an alcohol molecule forming {[LCoⅢ(SCN)]RCH2OH}2+ (Ⅱ) as observed by HR-ESI-MS (Fig. S8). Upon photo-excitation, alcohol provides an electron to the CoⅢ center and releases a proton, and then PCET from the activated alcohol to the coordinated azo-ligand occurred forming a monohydride intermediate species [HLCoⅡ(SCN)RCOH]+ (Ⅲ). This step is evidenced by the observation that a proton acceptor (tetrabutylammonium hydroxide or acetone) is necessary for the redox cycle. The intermediate species (Ⅲ) releases the generated aldehyde forming species Ⅳ, which can facilely donate the H atom on the hydrazine group to DBAD forming [LCoⅡ(SCN)(H-DBAD]+ (Ⅴ). After that, an electron transfers from the cobalt center to the H-DBAD under an acidic surrounding producing the corresponding hydrazine and [LCoⅢ(SCN)]2+ as evidenced by the fact that an azo ligand is necessary to oxidize the CoⅡ species. On the other hand, the species Ⅳ also provides a H atom to dioxygen and captures a SCN− anion generating complex 2 which is proven by the isolation of crystals of complex 2 by photo-irradiation of complex 1 in alcohol.
Scheme 1
Scheme 1.
Proposed mechanism of the cobalt switch driving alcohol dehydrogenation.
In conclusion, we have identified a pair of CoⅢ/CoⅡ redox switch which drives photo-dehydrogenation of alcohol. Stabilizing both the states of the redox pair provides some important insights into catalytic alcohol dehydrogenation. The concepts elucidated may provide an important foundation for future design of new redox catalyst systems for selective oxidation of organic molecules.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21871262, 21805275 and 21901242), the Natural Science Foundation of Fujian Province (No. 2019J01130), and the Recruitment Program of Global Youth Experts.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.06.053.
[1]
Z. Wu, Z. Zhen, J.H. Jiang, et al., Angew. Chem. Int. Ed. 56 (2017) 11060-11078. doi: 10.1002/anie.201702979
[2]
C. Tepper, G. Haberhauer, Antioxid. Redox Sign. 19 (2013) 1783-1791. doi: 10.1089/ars.2012.4740
[3]
T.X. Chen, F. Ning, H.S. Liu, et al., Chin. Chem. Lett. 28 (2017) 1380–1314.
Figure 3
Redox reaction driven mechanical motion of the Co switch. (a) Interconversion between two conformations of [LCoⅢ(SCN)]2+ (coplanar mode) and [LCoⅡ(SCN)]+ (saddle-like mode). (b) UV–vis absorption for photo-reduction (hv) of [LCoⅢ(SCN)]2+ and DBAD oxidation (ox) of [LCoⅡ(SCN)]+. (c) Reversible conversional cycles between the CoⅡ and CoⅢ species.

 DownLoad:
DownLoad:

 下载:
下载:





