Table 1.
Comparison of the training results from six different models.
Citation:
Jianzhong Zhu, Cheng Chen, Jie Dong, Shasha Cheng, Guodong Li, Chunming Wang, Defang Ouyang, Chung-Hang Leung, Ligen Lin. Artificial intelligence-aided discovery of prolyl hydroxylase 2 inhibitors to stabilize hypoxia inducible factor-1α and promote angiogenesis[J]. Chinese Chemical Letters,
2023, 34(2): 107514.
doi:
10.1016/j.cclet.2022.05.028

Artificial intelligence-aided discovery of prolyl hydroxylase 2 inhibitors to stabilize hypoxia inducible factor-1α and promote angiogenesis
English
Artificial intelligence-aided discovery of prolyl hydroxylase 2 inhibitors to stabilize hypoxia inducible factor-1α and promote angiogenesis
Abstract:
From ZINC database with a total of 1.8 million small molecules, four compounds are identified as prolyl hydroxylase 2 inhibitors through a virtual screening workflow that sequentially incorporates machine learning, molecular docking, and molecular dynamics. Among them, compound 103, (E)-5-(5-((2-(1H-tetrazol-5-yl)hydrazineylidene)methyl)furan-2-yl)isoindoline-1,3-dione, promotes the migration and capillary tube formation capacity of human umbilical vein endothelial cells through enhancing the stability of hypoxia inducible factor-1α and increasing the level of vascular endothelial growth factor.
-
Key words:
- Machine learning
- / Virtual screening
- / PHD2
- / Angiogenesis
- / Wound healing
-
Hypoxia-inducible factor (HIF)-1α is the determinant part of HIF-1 transcriptional activity [1]. Under normoxic condition, HIF-1α is naturally hydroxylated and degraded via the ubiquitin-mediated degradation pathway [2]. Whereas in hypoxia, HIF-1α escapes degradation, translocates to the nucleus and activates the transcription of a variety of angiogenic inducers such as vascular endothelial growth factor (VEGF) and fibroblast growth factor-2 [3]. The hydroxylation is catalysed by prolyl hydroxylases (PHDs), and PHD2 is found to be the critical oxygen sensor [4] and crucial in controlling HIF-1α levels. Dysfunctions of HIF-1α in diabetes have been indicated to deteriorate wound closure [5] and stabilization of HIF-1α has shown to improve wound healing [2, 3] and alleviate osteoarthritis [6]. Keratinocytes deficient in PHD2 stabilized HIF-1α and further induced expression of β3-integrin that promoted cell migration and improved wound healing [7]; enhanced wound healing was observed in diabetic mice after silencing PHD2 in fibroblast [3]; a cyclometalated iridium(Ⅲ) metal complex was recently reported to stabilize HIF-1α by occluding its binding to von Hippel-Lindau and succeeded in accelerating wound healing in db/db mice [8]. Therefore, PHD2 inhibitors have become attractive therapeutics for the treatment of diabetic foot.
Artificial intelligence (AI) has many applications in accelerating drug discovery process, such as sorting and classification of cells, prediction of physical properties of drugs, protein structure prediction, and drug-protein interaction prediction [9]. Machine learning is one of the tools currently studied most in computer-aided drug discovery [10]. Structure-based drug design is useful in discovering drug candidates and has become an important part in the discovery of new drugs [11]. It has been utilized to design inhibitors to protein targets like dipeptidyl peptidase 4 [12], sodium taurocholate cotransporting polypeptide [13] and Sirtuin 6 [14] with exhilarating results. While machine learning-based methods lose the 3D structure information of the ligand and protein, structure-based tools like molecular docking and molecular dynamics (MD) take good considerations of the chemical environment of the ligand binding sites of a target protein, the conformation of drug molecule, and the flexibility of a protein [9]. Therefore, in the present study, a virtual screening workflow sequentially incorporating machine learning, molecular docking and MD was performed to discover novel potential PHD2 inhibitors, aiming to stabilize HIF-1α and treat diabetic wound healing.
Descriptors and fingerprints of 1309 small molecules from BindingDB, containing 408 inhibitors and 901 non-inhibitors classified by a threshold of 1 µmol/L, were calculated to train 6 models. Principal component analysis was used to reduce dimensionality from 1646 to 350 with an explained variance ratio of 0.9977. Receiver operation curves were drawn and the area under the curve (AUC) was calculated (Fig. S1 in Supporting information). After evaluation and comparison (Table 1), support vector machine (SVM), with the highest accuracy, F1 score and outstanding AUC, was chosen as the best model for the first-step screening process.
Table 1
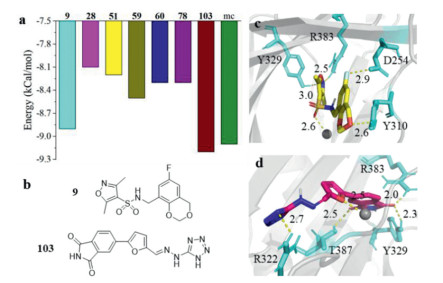
A total of 1.8 million anodyne and lead-like small molecules from ZINC were predicted with SVM afterwards. The resulting 402,067 molecules were firstly prepared under pH 7.4, filtered with Lipinski's 5 rules, and further screened with the rigid docking tool: LibDock. The successfully docked molecules were grouped into 1000 clusters. The best scored molecule in each cluster was taken and those owning high similarity (tanimoto coefficient > 0.20) to the known PHD2 ligands in the BindingDB database were excluded, leaving 124 molecules (numbered 1, 2, …, 124). These molecules were further docked by AutoDock Vina. The 17 molecules scored below −8.0 kcal/mol by Vina (Table S1 in Supporting information) had low similarity with each other (Fig. S2 in Supporting information) and were docked again with CDOCKER. The 4 molecules showing a root mean squared deviation lower than 2 Å to the conformation acquired in Vina plus the 3 highest scored molecules by Vina were chosen for further studies. The Vina docking score of the 7 molecules were displayed in Fig. 1a. The compound developed by Merck research laboratory (named mc afterwards), with an IC50 value of 0.2 nmol/L, was employed as a positive control [15]. Compounds N-((6-fluoro-4H-benzo[d][1,3]dioxin-8-yl)methyl)-3,5-dimethylisoxazole-4-sulfonamide (9, Fig. 1b) and (E)-5-(5-((2-(1H-tetrazol-5-yl)hydrazineylidene)methyl)furan-2-yl)isoindoline-1,3‑dione (103, Fig. 1b) haves scores comparable to that of mc, suggesting the great potentials to inhibit PHD2.
Figure 1
 Figure 1. The identification of PHD2 inhibitors from ZINC database through a virtual screening workflow. (a) Docking energy of compounds 9, 28, 51, 59, 60, 78, 103, and mc scored by AutoDock Vina. (b) The chemical structures of compounds 9 and 103. Interaction details of compounds 9 (c) and 103 (d) to PHD2 (PDB ID: 2HBT). Protein and molecules (residues) were shown in Cartoon and sticks, respectively; the Fe2+ was displayed in VDW mode with gray color; only residues owing hydrogen bonding, salt bridge, or coordinate bonding with the ligand were shown and residues showing hydrophobic interaction with the ligand were not displayed for clarity; yellow dotted line indicates hydrogen bond and the neighboring Arabic number is the length of the hydrogen bond.
Figure 1. The identification of PHD2 inhibitors from ZINC database through a virtual screening workflow. (a) Docking energy of compounds 9, 28, 51, 59, 60, 78, 103, and mc scored by AutoDock Vina. (b) The chemical structures of compounds 9 and 103. Interaction details of compounds 9 (c) and 103 (d) to PHD2 (PDB ID: 2HBT). Protein and molecules (residues) were shown in Cartoon and sticks, respectively; the Fe2+ was displayed in VDW mode with gray color; only residues owing hydrogen bonding, salt bridge, or coordinate bonding with the ligand were shown and residues showing hydrophobic interaction with the ligand were not displayed for clarity; yellow dotted line indicates hydrogen bond and the neighboring Arabic number is the length of the hydrogen bond.Interaction was scrutinized using compounds 9 and 103 as representatives as they were the highest scored molecules by Vina. Like most available PHD2 inhibitors of carboxylic acid derivatives, compounds 9 and 103 were found to form salt bridge or hydrogen bonds with Y329 and R383, and coordinate bond with the Fe2+ ion. Specifically, for compound 9 (Fig. 1c and Table S2 in Supporting information), the five-member ring lied inside the pocket and was in close proximity to Y329 and R383, with the nitrogen atom forming one hydrogen bond each with the two residues; one halogen bond was formed between D254 and the fluorine atom; the polar hydrogen on the sidechain of Y310 was hydrogen bonded with the oxygen atom (2.6 Å); the sulfur atom displayed π-sulfur interaction with H314 and H374; additional hydrophobic interactions with M299, A301, Y303, I327, V376 and A385 were also detected. Similarly, the isoindoline-1,3-dione group of compound 103 was immersed in the pocket, whereas the tetrazole ring situated at the entrance of the pocket and pointed obliquely outwards (Fig. 1d and Table S2): The carbonyl oxygen was hydrogen bonded with Y329 (2.3 Å) and R383 (2.0 Å), and the other oxygen atom on the other side of the ring further formed a hydrogen bond with T387 (2.5 Å); π-cation electrostatic interaction was also found between the five-member ring and R383; the whole molecule was further anchored by a coordinate bond formed between Fe2+ and the oxygen atom on the furan ring as well as a hydrogen bond formed with R322 at the other end; 103 also exhibited extra hydrophobic interaction with Y310, I327, A385, and W389. Therefore, the binding mode of 9 and 103 retained the ionic interaction with R383 and coordination with ferrous iron, two key pharmacophores in 2-oxogluarate derivatives [16]. Docking conformations of the other molecules and interaction summary were shown in Fig. S3 (Supporting information) and Table S2.
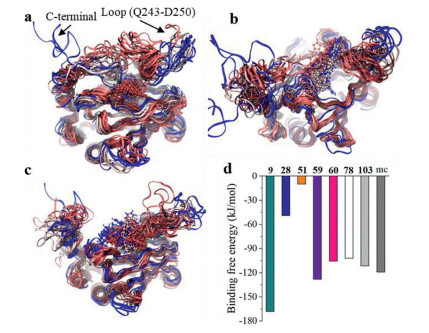
To evaluate the binding affinity more accurately, MD was performed. The trajectories of compounds 9 (Fig. 2a), 103 (Fig. 2b), and mc (Fig. 2c) were selected as representatives, and that of the others were shown in Fig. S4 (Supporting information). All the molecules except 78 stayed in the active pocket in the 300 ns long simulation. Compound 78 stayed at the pocket for the first 60 ns, then moved out of the pocket and interacted with an α-helix at the C-terminal and a nearby loop (Fig. S4e), and further bound to another loop below the C-terminal. For compound 9, the random coil from the C-terminal and the loop (Q243‒D250) were found to bend towards the pocket center, forming interaction with the small molecule and reducing the possibility that the compounds moved away from the pocket (Fig. 2a). This was also observed for compounds 51 (Fig. S4b) and 60 (Fig. S4d). Although the isoindoline-1, 3‑dione group moiety of 103 that inserted deep into the pocket moved outwards at the first nanosecond, it was stabilized for the remaining 299 ns with an obliquely upwards conformation across the channel entrance: one end was anchored with a β-sheet and a random coil above the pocket and the other end was anchored with another coil at the entrance of the pocket (Fig. 2b). We believed that this stable binding conformation could effectively occlude the entrance of further substrates. The tail of mc vacillated back and forth, but the carboxyl end remained immersed in the pocket for the whole trajectory due to the strong electrostatic interaction (hydrogen bonding and salt bridge) and hydrophobic interaction in the central moiety as well (Fig. 2c). Binding free energy of all the molecules (Fig. 2d) was estimated from the stable trajectory: compounds 9, 59, 60, 78 and 103 possessed comparable values to that of mc. Due to the fact that 78 bound to a site far away from the pocket as discussed above, it was excluded for further study. Therefore, compounds 9, 59, 60 and 103 were chosen for in vitro studies.
Figure 2
 Figure 2. Trajectory of the protein-compound complex (a: 9, b: 103, c: mc). Binding free energy of the 8 compounds estimated by g_mmpbsa (d). Protein and small molecules were shown in Newribbons and CPK, respectively, and were colored as timestep in blue-white-red scale; blue, white, and red color indicate earlier, middle, recent frames in the trajectory, respectively.
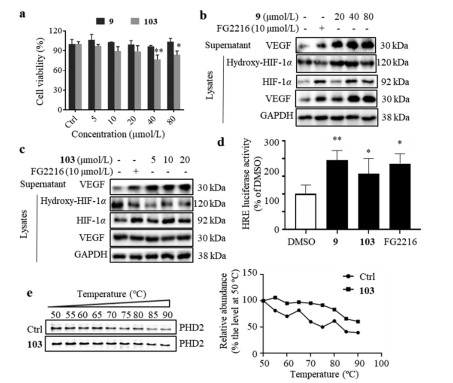
Figure 2. Trajectory of the protein-compound complex (a: 9, b: 103, c: mc). Binding free energy of the 8 compounds estimated by g_mmpbsa (d). Protein and small molecules were shown in Newribbons and CPK, respectively, and were colored as timestep in blue-white-red scale; blue, white, and red color indicate earlier, middle, recent frames in the trajectory, respectively.Compounds 9 and 103 were commercially available, which were characterized with their 1H and 13C NMR spectra (Figs. S5‒S8 in Supporting information). Compound 9 was not cytotoxic to human umbilical vein endothelial cells (HUVECs) up to 80 µmol/L, while the maximum safe dose of compound 103 was 20 µmol/L (Fig. 3a). The protein levels of hydroxy‑HIF-1α, total HIF-1α and VEGF in cell lysates, as well as the protein level of VEGF in culture medium from HUVECs treated by compound 9 or 103 were firstly determined by Western blots. Like FG2216, treatment of compound 103 decreased hydroxy‑HIF-1α level and increased total HIF-1α level in cell lysate, and increased secretory VEGF in culture medium; and treatment of compound 9 did not change hydroxy‑HIF-1α level, but increased total HIF-1α and VEGF levels (Figs. 3b and c). The dual-luciferase reporter assay was further performed on human embryonic kidney (HEK)−293T cells to monitor the hypoxia response element (HRE)-driven luciferase activity induced by compound 9 or 103. After the cells were treated with compound 9 or 103 (80 µmol/L) for 24 h, the HRE-driven luciferase activity was significantly increased, which was comparable with the positive control, FG2216 (Fig. 3d). As a valid method, cellular thermal shift assay (CETSA) was performed to explore whether compound 103 interacts with PHD2 [13]. Compared with the control, the treatment of compound 103 obviously stabilized PHD2 at various temperatures, which indicated that compound 103 might bind to PHD2 to increase the thermal stability (Fig. 3e).
Figure 3
 Figure 3. Compounds 9 and 103 enhanced the stability of HIF-1α. (a) Cytotoxicity of compounds 9 and 103 on HUVECs assessed by MTT assay. HUVECs were treated with compounds for 24 h. The protein levels of hydroxy‑HIF-1α, total HIF-1α and VEGF in the cell lysates, and the protein level of VEGF in the culture medium of HUVECs treated with compound 9 (b) or 103 (c). HUVECs were treated with different concentrations of compound 9 or 103 for 24 h; Roxadustat (FG2216, a PHD2 inhibitor) was employed as a positive control; GAPDH was chosen as an internal loading control. (d) Effect of compounds 9 and 103 on HIF-1α activation of the HRE-driven reporter on HEK-293T cells. FG2216 was used as a positive control. HEK-293T cells were treated with compound 9 (80 µmol/L), 103 (80 µmol/L) or FG2216 (80 µmol/L) for 24 h. n = 3. (e) CETSA was performed on HUVECs treated with or without compound 103 (20 µmol/L) for 24 h. n = 3. Data was expressed as mean ± SEM from at least three individual experiments. *P < 0.05 and **P < 0.01, versus DMSO.
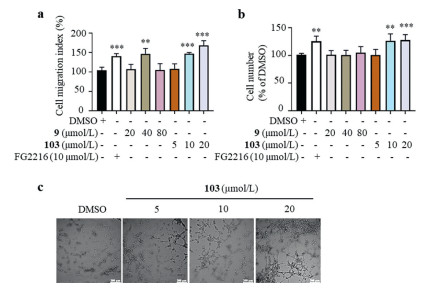
Figure 3. Compounds 9 and 103 enhanced the stability of HIF-1α. (a) Cytotoxicity of compounds 9 and 103 on HUVECs assessed by MTT assay. HUVECs were treated with compounds for 24 h. The protein levels of hydroxy‑HIF-1α, total HIF-1α and VEGF in the cell lysates, and the protein level of VEGF in the culture medium of HUVECs treated with compound 9 (b) or 103 (c). HUVECs were treated with different concentrations of compound 9 or 103 for 24 h; Roxadustat (FG2216, a PHD2 inhibitor) was employed as a positive control; GAPDH was chosen as an internal loading control. (d) Effect of compounds 9 and 103 on HIF-1α activation of the HRE-driven reporter on HEK-293T cells. FG2216 was used as a positive control. HEK-293T cells were treated with compound 9 (80 µmol/L), 103 (80 µmol/L) or FG2216 (80 µmol/L) for 24 h. n = 3. (e) CETSA was performed on HUVECs treated with or without compound 103 (20 µmol/L) for 24 h. n = 3. Data was expressed as mean ± SEM from at least three individual experiments. *P < 0.05 and **P < 0.01, versus DMSO.These results prompted us to investigate the pro-angiogenic effects of both compounds on HUVECs. In scratch assay [17], both compounds 9 and 103 enhanced the mobility of HUVECs (Fig. 4a); in transwell assay [18], compound 103 but not compound 9 increased the migration ability of HUVECs in a dose-dependent manner (Fig. 4b). Thus, compound 103 was chosen for further tube formation assay [19, 20]. There was rare tube formation when HUVECs were incubated on Matrigel in low serum medium, whereas obvious tube formation was induced when HUVECs were pre-treated with different concentrations of compound 103 for 5 h (Fig. 4c). Quantitative analysis of key parameters in tube formation experiment (the number of nodes and master segments, total master segments length and total length) showed that compound 103 promoted angiogenesis when compared with the vehicle cells (Fig. S9 in Supporting information). The wound healing process includes three phases: inflammation, new tissue formation, and remodeling; the second phase relates to the proliferation and migration of lots of different cell types [21]. Tissue formation requires new blood vessel growth through angiogenesis or vasculogenesis so that vasculature can be re-established to secure the supply of oxygen and nutrients [22]. The pro-angiogenic effect of compound 103 on HUVECs indicates its application prospects in accelerating diabetic wound healing.
Figure 4
 Figure 4. Compound 103 enhanced the migration and capillary tube formation capacity of HUVECs. HUVECs were treated with different compounds at various concentrations for specific time (24 h for scratch assay and 6 h for transwell migration assay). The effects of compounds 9 and 103 on the mobility of HUVECs, assessed by scratch assay (a) and transwell migration assay (b). (c) Compound 103 promoted tube formation capacity of HUVECs. Representative images of tube formation of HUVECs treated with different concentrations of compound 103 for 5 h. Scale bar = 200 µm. n = 3. Data was expressed as mean ± SEM from at least three individual experiments. *P < 0.05, **P < 0.01, and ***P < 0.001, versus DMSO.
Figure 4. Compound 103 enhanced the migration and capillary tube formation capacity of HUVECs. HUVECs were treated with different compounds at various concentrations for specific time (24 h for scratch assay and 6 h for transwell migration assay). The effects of compounds 9 and 103 on the mobility of HUVECs, assessed by scratch assay (a) and transwell migration assay (b). (c) Compound 103 promoted tube formation capacity of HUVECs. Representative images of tube formation of HUVECs treated with different concentrations of compound 103 for 5 h. Scale bar = 200 µm. n = 3. Data was expressed as mean ± SEM from at least three individual experiments. *P < 0.05, **P < 0.01, and ***P < 0.001, versus DMSO.To conclude, compound 103 was identified as a PHD2 inhibitor with novel scaffold through a virtual screening workflow (machine learning followed by molecular docking and MD). Compound 103 was able to stabilize HIF-1α, increase the transcriptional activity of HIF-1α, and enhance the migration ability of HUVECs. Aqueous solubility is essential for drug candidates. As predicted on ADMETlab 2.0, all the scoring terms of compound 103 are located well into the buffer zone composed by the upper limit and lower limit (Fig. S10 in Supporting information). The aqueous solubility of compound 103 is about 2.78 g/L at room temperature, and the n-octanol-water partition coefficient LogP is about 0.98. Thus, structural modification or drug carrier is needed to be investigated in the future. Compound 103 is worthy further investigation and development as a therapeutic agent to treat diabetic foot ulcers.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This work is financially supported by the National Natural Science Foundation of China (Nos. 82073715, 81872754), the Science and Technology Development Fund, Macau SAR (No. FDCT 0001/2021/AKP), and the Research Fund of University of Macau (No. MYRG2020-00091-ICMS).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:
10.1016/j.cclet.2022.05.028 .
-
-
[1]
G.L. Semenza, Cell 148 (2012) 399–408. doi: 10.1016/j.cell.2012.01.021
-
[2]
I.R. Botusan, V.G. Sunkari, O. Savu, et al., Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 19426–19431. doi: 10.1073/pnas.0805230105
-
[3]
X. Zhang, X. Yan, L. Cheng, et al., PLoS One 8 (2013) e84548. doi: 10.1371/journal.pone.0084548
-
[4]
M.K. Teli, S. Kumar, D.K. Yadav, M.H. Kim, J. Biomol. Struct. Dyn. 39 (2021) 703–717. doi: 10.1080/07391102.2020.1714480
-
[5]
H. Thangarajah, I.N. Vial, R.H. Grogan, et al., Cell Cycle 9 (2010) 75–79. doi: 10.4161/cc.9.1.10371
-
[6]
S. Hu, C. Zhang, L. Ni, et al., Cell Death Dis. 11 (2020) 481. doi: 10.1038/s41419-020-2680-0
-
[7]
J. Kalucka, A. Ettinger, K. Franke, et al., Mol. Cell. Biol. 33 (2013) 3426–3438. doi: 10.1128/MCB.00609-13
-
[8]
G. Li, C.N. Ko, D. Li, et al., Nat. Commun. 12 (2021) 3363. doi: 10.1038/s41467-021-23448-7
-
[9]
H.C.S. Chan, H. Shan, T. Dahoun, H. Vogel, S. Yuan, Trends Pharmacol. Sci. 40 (2019) 592–604. doi: 10.1016/j.tips.2019.06.004
-
[10]
J. Du, J. Guo, D. Kang, et al., Chin. Chem. Lett. 31 (2020) 1695–1708. doi: 10.1016/j.cclet.2020.03.028
-
[11]
Z. Yu, Z. Li, Q. Yu, et al., Chem. Biol. Drug Des. 95 (2020) 270–278. doi: 10.1111/cbdd.13640
-
[12]
S. Li, C. Qin, S. Cui, et al., J. Med. Chem. 62 (2019) 2348–2361. doi: 10.1021/acs.jmedchem.8b01491
-
[13]
H. Xiang, Y. Chen, J. Zhang, et al., Chin. Chem. Lett. 31 (2020) 1422–1426. doi: 10.1016/j.cclet.2020.03.017
-
[14]
Z. Huang, J. Zhao, W. Deng, et al., Nat. Chem. Biol. 14 (2018) 1118–1126. doi: 10.1038/s41589-018-0150-0
-
[15]
P. Vachal, S. Miao, J.M. Pierce, et al., J. Med. Chem. 55 (2012) 2945–2959. doi: 10.1021/jm201542d
-
[16]
Y. Wu, N. Wang, Y. Lei, et al., Med. Chem. Comm. 7 (2016) 1271–1284. doi: 10.1039/C6MD00240D
-
[17]
J. Huang, X. You, P. Xin, et al., Chin. Chem. Lett. 32 (2021) 1737–1742. doi: 10.1016/j.cclet.2020.12.006
-
[18]
J. Fan, Y. Fan, Z. Wei, et al., Chin. Chem. Lett. 31 (2020) 1787–1791. doi: 10.1016/j.cclet.2020.03.065
-
[19]
S. Wen, K. Zhang, Y. Li, et al., Chin. Chem. Lett. 31 (2020) 3153–3157. doi: 10.1016/j.cclet.2020.03.077
-
[20]
R. Lou, F. Xu, Y. Xu, et al., Bioorg. Chem. 117 (2021) 105426. doi: 10.1016/j.bioorg.2021.105426
-
[21]
G.C. Gurtner, S. Werner, Y. Barrandon, M.T. Longaker, Nature 453 (2008) 314–321. doi: 10.1038/nature07039
-
[22]
R.J. Ruthenborg, J.J. Ban, A. Wazir, N. Takeda, J.W. Kim, Mol. Cells 37 (2014) 637–643. doi: 10.14348/molcells.2014.0150
-
[1]
-
Figure 1 The identification of PHD2 inhibitors from ZINC database through a virtual screening workflow. (a) Docking energy of compounds 9, 28, 51, 59, 60, 78, 103, and mc scored by AutoDock Vina. (b) The chemical structures of compounds 9 and 103. Interaction details of compounds 9 (c) and 103 (d) to PHD2 (PDB ID: 2HBT). Protein and molecules (residues) were shown in Cartoon and sticks, respectively; the Fe2+ was displayed in VDW mode with gray color; only residues owing hydrogen bonding, salt bridge, or coordinate bonding with the ligand were shown and residues showing hydrophobic interaction with the ligand were not displayed for clarity; yellow dotted line indicates hydrogen bond and the neighboring Arabic number is the length of the hydrogen bond.
Figure 2 Trajectory of the protein-compound complex (a: 9, b: 103, c: mc). Binding free energy of the 8 compounds estimated by g_mmpbsa (d). Protein and small molecules were shown in Newribbons and CPK, respectively, and were colored as timestep in blue-white-red scale; blue, white, and red color indicate earlier, middle, recent frames in the trajectory, respectively.
Figure 3 Compounds 9 and 103 enhanced the stability of HIF-1α. (a) Cytotoxicity of compounds 9 and 103 on HUVECs assessed by MTT assay. HUVECs were treated with compounds for 24 h. The protein levels of hydroxy‑HIF-1α, total HIF-1α and VEGF in the cell lysates, and the protein level of VEGF in the culture medium of HUVECs treated with compound 9 (b) or 103 (c). HUVECs were treated with different concentrations of compound 9 or 103 for 24 h; Roxadustat (FG2216, a PHD2 inhibitor) was employed as a positive control; GAPDH was chosen as an internal loading control. (d) Effect of compounds 9 and 103 on HIF-1α activation of the HRE-driven reporter on HEK-293T cells. FG2216 was used as a positive control. HEK-293T cells were treated with compound 9 (80 µmol/L), 103 (80 µmol/L) or FG2216 (80 µmol/L) for 24 h. n = 3. (e) CETSA was performed on HUVECs treated with or without compound 103 (20 µmol/L) for 24 h. n = 3. Data was expressed as mean ± SEM from at least three individual experiments. *P < 0.05 and **P < 0.01, versus DMSO.
Figure 4 Compound 103 enhanced the migration and capillary tube formation capacity of HUVECs. HUVECs were treated with different compounds at various concentrations for specific time (24 h for scratch assay and 6 h for transwell migration assay). The effects of compounds 9 and 103 on the mobility of HUVECs, assessed by scratch assay (a) and transwell migration assay (b). (c) Compound 103 promoted tube formation capacity of HUVECs. Representative images of tube formation of HUVECs treated with different concentrations of compound 103 for 5 h. Scale bar = 200 µm. n = 3. Data was expressed as mean ± SEM from at least three individual experiments. *P < 0.05, **P < 0.01, and ***P < 0.001, versus DMSO.
-

 DownLoad:
DownLoad:


 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1739
- HTML全文浏览量: 203
