
Scheme 1.
Previous work and this reaction.
Construction of chiral 3-alkenyl-3-substituted oxindoles by stereoselective direct alkenylation of isatin derivatives and 3-vinylindoles
Xiang Sun , Kuiliang Li , Shuangshuang Zhao , Zhenggen Zha , Zhiyong Wang

3-Substituted 2-oxindole is one of the most abundant and significant structural motifs in natural products [1-3]. They are widely found in biologically active molecules and drugs. For example, spirotryprostatin B alkaloids exhibit cytotoxic activity [4, 5]. The JP-8g proved to be potential for cancer treatment and inflammation therapy [6]. AG041R is a gastrin/CCK-B receptor agonist [7], and NITD609 is an excellent antimalarial drug candidate [8-11]. The C3 substituent and absolute configuration were the key points for 3-substituted 2-oxindoles biologically activity. Therefore, the construction of chiral center at the C3 position of oxindoles has attracted extensive attention in chemistry.
Isatin, a well-known natural product found in plants of genus Isatis and Couroupita guianensis aubl [12, 13] has been widely used in synthetic chemistry [14-18]. To the best of our knowledge, the asymmetric alkenylation of isatins was rarely reported. In 2010, Zhou's group reported the asymmetric synthesis of isatins with acrolein catalyzed by cinchona alkaloid and high yields and enantioselectivities were obtained (Scheme 1a) [19]. In 2016, Zhao's group reported the asymmetric alkenylation reaction of isatins with alkenyl boronic acid catalyzed by chiral CoI2-bisphosphine complex at 70 ℃ and the desired product was obtained with 93% ee (Scheme 1b) [20]. However, appropriate substituents are needed at the terminal site of the alkenyl groups. Up to now, some asymmetric Friedel-Crafts-type olefination reaction of ketones and ketimines [21, 22] were reported, but we have not found the direct alkenylation of isatin by the Friedel-Crafts reaction. Based on our previous Lewis acid catalytic system on the Friedel-Crafts reaction [23-30] of aromatic rings, we intend to study the asymmetric direct alkenylation of isatin. Recently, 3-vinylindoles and derivatives have emerged as a kind of versatile chemical synthon in asymmetric transformations [31-36], Guo's group made great contributions to the research of 3–vinylindoles (Scheme 1c). Herein we realized a highly asymmetric direct alkenylation of isatins with 3–vinylindoles by virtue of our chiral copper complex [37, 38]. A variety of chiral 3-alkenyl 3-substituted oxindoles (Scheme 1d) could be obtained with excellent yields (up to 99%) and ee values (up to 99%). This is an alternative asymmetric reaction for the 3–vinylindoles and the isatins.
We initiated our investigation with the model reaction between 1a and 2a by virtue of our chiral copper complex. Firstly, different temperatures and copper salts were screened for this reaction (see section 1.2 in Supporting information). It was found that 20 ℃ was the best temperature and CuBr2 was the best Lewis acid for this reaction, affording the corresponding product with 87% yield and 82% ee. Next, various solvents were examined in this reaction. It was found that ethanol was the best solvent (Table 1, entries 1-5). Subsequently, the electronic effect and the steric effect of the ligands were investigated (Table 1, entries 1, 6-10). Neither strong electron withdrawing substituent CF3 at the phenyl of ligand (L) nor the strong electron donating substituent OCH3 at the phenyl of ligand (L) favored this reaction. The commercial Box Ligand L could not improve the enantioselectivity either. Even worse, L did not work in this reaction. Finally, we found that L was still the most efficient ligand. With the optimal L-Cu catalyst in hand, various bases (Table 1, entries 1, 11-15) were examined. The employment of N-ethylmorpholine gave the best results (Table 1, entry 14). To further improve the enantioselectivity, the dosage of N-ethylmorpholine was optimized (Table 1, entries 14, 16-19). It was found that 2 equiv. N-Ethylmorpholine gave the corresponding product with 89% yield, 91% ee. In summary, the optimal reaction conditions were listed as follows: the reaction being carried out in ethanol at 20 ℃ for 36 h with L as the ligand, CuBr2 as the Lewis acid, ethanol as the solvent and two equivalent N-ethylmorpholine as the base.
Under the optimized reaction conditions, the substrate scope was then explored. The results were summarized in Scheme 2. First of all, the substrates bearing different protective group on the N atom of isatin was employed. We found that the reaction had a great substrate compatibility. For instance, the substrates bearing the benzyl, phenyl and allyl group at the N atom of the isatins can be employed in the reaction to give the corresponding products with good yields and enantioselectivities (4a-4c, Scheme 2). Furthermore, we studied the electronic effects by changing the substitution at the para position on N-benyl ring of isatins (4d-4g, Scheme 2). We found that the reactions can be carried out smoothly to afford the corresponding products with high yields and good ee values regardless of the electron-withdrawing or electron-donating substituents on the N atom. This indicated that electronic effect had little influence on the reaction. Afterwards, different substituted 3-(1-phenyl-vinyl)-indoles were then investigated. When the bromo groups or methoxyl groups were installed at 5- and 6-position of the indole ring of the 3-(1-arylvinyl)-1H-indoles, the corresponding products 4h was generated in excellent diastereoselectivities, with a little reduced yields and enantioselectivities, while 4i was generated in excellent ee and dr values. When the electron-donating methoxyl group were installed at 3- and 4-position of the phenyl ring, the desired products could be obtained with high diastereo- and enantioselectivities (4k and 4m, Scheme 2). When the methyl and fluoro groups were installed at 4-position of the phenyl ring, the desired products can be obtained with a slightly reduced yields and ee values while the dr values can be maintained (4l and 4n, Scheme 2). This implied that the electronic effect from substrate 3-(1-phenyl-vinyl)-indoles had little influence on the reaction. Furthermore, various substituted isatins were then employed as the electrophiles in the reactions. Isatins bearing electron-donating methyl and methoxyl groups at the C5 position gave the desired product 4o, 4p in excellent dr and ee values. In contrast, when halogen chloro and bromo substituents at different positions of isatin, the high yields and excellent dr values can be obtained in spite of a little decrease in the ee values (4q-4u, Scheme 2). It was noted that the addition of a methyl group at C4 position can enhance both the yield and ee value to some extent (4v, Scheme 2). This meant that electron donation substituents at the isatin ring favored this reaction.
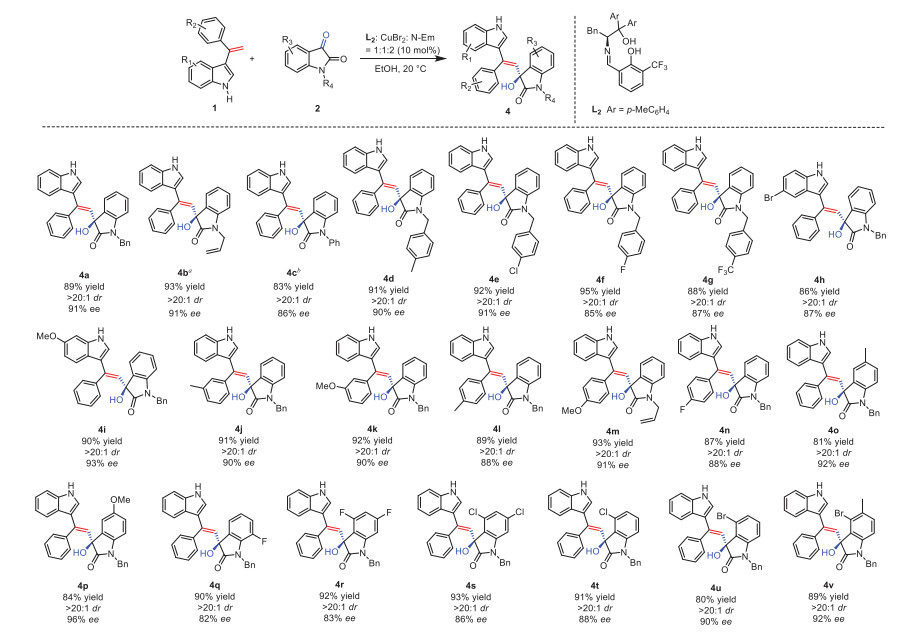
On the other hand, the substrate isatins can be extended to the isatin imines under the standard condition when the reaction temperature was modulated to 15 ℃, as shown in Scheme 3. We found that 3-vinylindoles with different substituents on the phenyl ring can be employed in this reaction to afford the corresponding products with both high yields and excellent ee values regardless of electron-donating or electron-withdrawing substituents on the para or meta position of the phenyl ring. For instance, the phenyl ring bearing a strong electron-withdrawing trifluoromethyl group, the corresponding product 5g can be obtained with high yield (90%) and excellent stereoselectivities (dr 20:1, 99% ee). When the phenyl ring bearing an electron-donation methoxyl group, in contrast, the reaction can also afford the corresponding product 5b with a yield of 99% and excellent stereoselectivities (dr 20:1, 97% ee). This meant that the reaction can carried out smoothly regardless of the electronic effect. Being different from the isatins, the alkylation of the NH in the indole ring of the isatin imines had little influence on the asymmetric alkenylation. For instance, when the methyl group was installed at 1-position of 3-vinylindoles, the corresponding product 5j can be obtained in a yield of 93% with 97% ee and > 20:1 dr. Similarly 5k can be obtained in a yield of 95% with 93% ee and > 20:1 dr. Nevertheless, the reaction time should be prolonged to 36 h and the reaction temperature should be increased to 25 ℃.
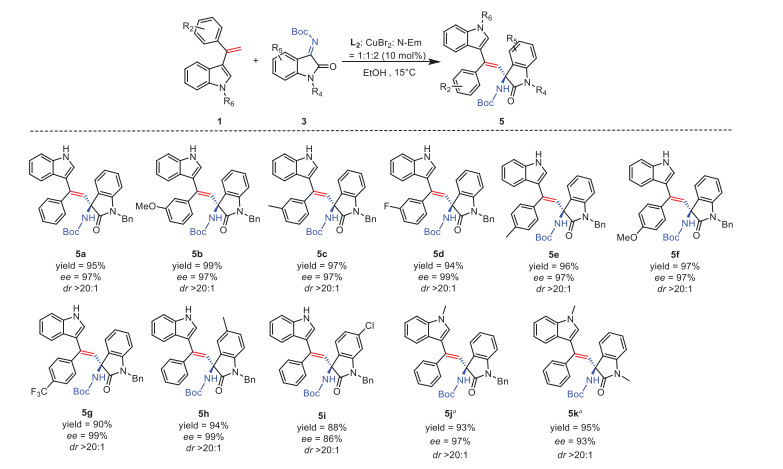
In order to gain insight into the reaction mechanism, some control experiments were performed (Scheme 4). When the phenyl group in 3–(1-phenyl-vinyl)-indole was replaced with a hydrogen (Scheme 4A) or a methyl group (Scheme 4B), the reaction did not work. In terms of this experimental result, we proposed a possible π-π interaction in the reaction mechanism. On the other hand, this mechanism was supported by DFT calculations (for details of DFT calculations and references see Supporting information 1.6). In the presence of 2 equiv. N-ethylmorpholine, Int-Ⅰ is generated from CuBr2 and the ligand L. Coordination of Int-Ⅰ with 1a and 2a affords Int-Ⅱ. The π-π interaction between 3-(1-phenyl-vinyl)-indole and isatin stabilizes the Int-Ⅱ structure. The 3-(1-phenyl-vinyl)-indole attacks from the Si-face due to the steric hindrance of trifluoromethylbenzene on the ligand. Next, the resulting Int-Ⅲ undergoes ligand exchange, proton shift and dissociation to give the final product. The 3D structure of transition state TS is also demonstrated in Scheme 4C.
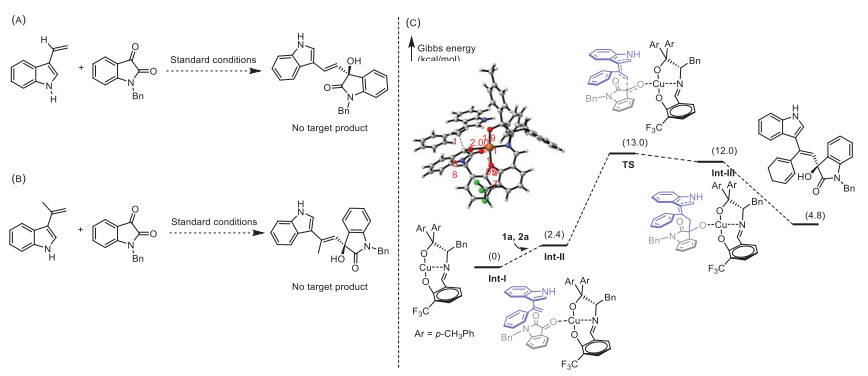
To evaluate the practicability of this new alkenylation reaction, a gram scale of experiment was performed under the standard condition. As shown in Scheme 5, 1.79 g of 4b could be obtained with 88% yield, 87% ee and > 20:1 dr in the presence of 10 mol% catalyst. The allyl alcohol structure in 3-alkenyl-3-substituted oxyindoles could be readily converted to other β-substituted ketones [40-42] or allyl sulfides [43]. Therefore these β-substituted ketones and allyl sulfides have potential synthetic utility in pharmaceutical industry since many biological molecules contain these moieties.
In summary, we developed an asymmetric alkenylation reaction of isatin derivatives and 3-vinylindoles. A series of chiral 3-alkenyl-3-substituted oxindole derivatives in gram scale were obtained with excellent yields and stereoselectivities. Also, the reaction transition state with a π-π interaction was proposed, which was supported by DFT calculations. Notably, the products are rich in functional group conversion and have great potential application for the synthesis of some natural products and drug molecules.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We are grateful for the financial support from the National Natural Science Foundation of China (No. 21772185). This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB20000000).
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.04.032.
S. Mohammadi, R. Heiran, R.P. Herrera, et al., ChemCatChem 5 (2013) 2131–2148. doi: 10.1002/cctc.201300050
G.S. Singh, Z.Y. Desta, Chem. Rev. 112 (2012) 6104–6155. doi: 10.1021/cr300135y
F. Zhou, Y. Liu, J. Zhou, Adv. Synth. Catal. 352 (2010) 1381–1407. doi: 10.1002/adsc.201000161
F.V. Nussbaum, S.J. Danishefsky, Angew. Chem. Int. Ed. 39 (2000) 2175–2178. doi: 10.1002/1521-3773(20000616)39:12<2175::AID-ANIE2175>3.0.CO;2-J
X. Jiang, Y. Sun, J. Yao, et al., Adv. Synth. Catal. 354 (2012) 917–925. doi: 10.1002/adsc.201100792
Y. Sun, J. Liu, T. Sun, et al., Sci. Rep. 4 (2014) 4372.
M. Ochi, K. Kawasaki, H. Kataoka, et al., Biochem. Biophys. Res. Commun. 283 (2001) 1118–1123. doi: 10.1006/bbrc.2001.4911
J.C. Pelt-Koops, H.E. Pett, W. Graumans, et al., Antimicrob. Agents Chemother. 56 (2012) 3544–3548. doi: 10.1128/AAC.06377-11
X. Liu, H. Zheng, C. Xu, et al., Angew. Chem. Int. Ed. 54 (2015) 10958–10962. doi: 10.1002/anie.201505717
H. Turner, Futur. Med. Chem. 8 (2016) 227–238. doi: 10.4155/fmc.15.177
J.F.M. da Silva, S. J Garden, A. C Pinto, J. Braz. Chem. Soc. 12 (2001) 273. doi: 10.1590/S0103-50532001000300002
J. Bergman, J.O. Lindstrom, U. Tilstam, Tetrahedron 41 (1988) 2879–2881. doi: 10.1016/s0040-4020(01)96609-8
Q. Chen, Y. Tang, T. Huang, et al., Angew. Chem. Int. Ed. 55 (2016) 5286–5289. doi: 10.1002/anie.201600711
G. Wang, X. Liu, Y. Chen, et al., ACS Catal. 6 (2016) 2482–2486. doi: 10.1021/acscatal.6b00294
J. Xu, J. Peng, C He, H. Ren, Org. Chem. Front. 6 (2019) 172–176. doi: 10.1039/C8QO01085D
J. Wang, Q Zhang, Y Li, et al., Chem. Commun. 56 (2020) 261–264. doi: 10.1039/C9CC07944K
J. Duan, Y. Mao, A. Xian, et al., Chem. Commun. 57 (2021) 3379–3382. doi: 10.1039/D0CC07995B
J. Li, Y. Li, J. Sun, et al., Chem. Commun. 55 (2019) 6309–6312. doi: 10.1039/C9CC02159K
Y. Liu, B. Wang, J. Cao, et al., J. Am. Chem. Soc. 132 (2010) 15176–15178. doi: 10.1021/ja107858z
Y. Huang, R. Huang, Y. Zhao, J. Am. Chem. Soc. 138 (2016) 6571–6576. doi: 10.1021/jacs.6b02372
B. Xiang, T.F. Xu, L. Wu, et al., J. Org. Chem. 81 (2016) 3929–3935. doi: 10.1021/acs.joc.6b00358
R.R. Liu, D.J. Wang, L. Wu, et al., ACS Catal. 5 (2015) 6524–6528. doi: 10.1021/acscatal.5b01793
K. Li, X. Sun, L. Li, et al., Chem. Eur. J. 27 (2021) 581–584. doi: 10.1002/chem.202003510
F. Guo, G. Lai, S. Xiong, et al., Chem. Eur. J. 16 (2010) 6438–6441. doi: 10.1002/chem.201000540
C. Li, F. Guo, K. Xu, et al., Org. Lett. 16 (2014) 3192–3195. doi: 10.1021/ol501086q
J. Sun, Y. Hu, Y. Li, et al., J. Org. Chem. 82 (2017) 5102–5110. doi: 10.1021/acs.joc.7b00159
Y. Gui, Y. Li, J. Sun, et al., J. Org. Chem. 83 (2018) 7491–7499. doi: 10.1021/acs.joc.8b01141
J. Sun, Y. Gui, Y. Huang, et al., ACS Omega 5 (2020) 11962–11970. doi: 10.1021/acsomega.9b04115
X. Liang, Y. Gui, K. Li, et al., Chem. Commun. 56 (2020) 11118–11121. doi: 10.1039/D0CC04410E
X. Li, W. Yuan, S. Tang, et al., Org. Lett. 19 (2017) 1120–1123. doi: 10.1021/acs.orglett.7b00143
X. Li, W. Hu, Q. Xiong, et al., Adv. Synth. Catal. 361 (2019) 1803–1807. doi: 10.1002/adsc.201801705
Y. Huang, X. Li, L. Fu, Q. Guo, Org. Lett. 18 (2016) 6200–6203. doi: 10.1021/acs.orglett.6b03257
W. Hu, X. Li, W. Gui, et al., Org. Lett. 21 (2019) 10090–10093. doi: 10.1021/acs.orglett.9b04063
X. Guan, H. Zhang, J. Gao, et al., J. Org. Chem. 84 (2019) 12562–12572. doi: 10.1021/acs.joc.9b02024
J.H. Xue, M. Shi, F. Yu, et al., Org. Lett. 18 (2016) 3874–3877. doi: 10.1021/acs.orglett.6b01880
Y.W. Sun, Z.S. Wang, S.F. Wu, Y.C. Zhang, F. Shi, Green Synth. Catal. 3 (2022) 84–88. doi: 10.1016/j.gresc.2021.11.003
G.Y. Lai, S.J. Wang, Z.Y. Wang, Tetrahedron: Asymmetry 19 (2008) 1813–1819. doi: 10.1016/j.tetasy.2008.06.036
F. Guo, D. Chang, G. Lai, et al., Chem. Eur. J. 17 (2011) 11127–11130. doi: 10.1002/chem.201102206
C. Y. Legault, C.Y. Lview, 1.0b, Universit de Sherbrooke, 2009, http://www.cylview.org.
Z. Guan, H. Wang, Y. Huang, et al., Org. Lett. 21 (2019) 4619–4622. doi: 10.1021/acs.orglett.9b01518
Z. Chen, W. Bai, S. Wang, et al., Angew. Chem. Int. Ed. 52 (2013) 9781–9785. doi: 10.1002/anie.201304557
Z. Zeng, X Xun, L Huang, et al., Green. Chem. 20 (2018) 2477–2480. doi: 10.1039/C8GC00478A
G. Guo, Y. Yuan, S. Wan, et al., Org. Chem. Front. 8 (2021) 2990–2996. doi: 10.1039/D1QO00148E

Scheme 2 Scope of substrates. Unless otherwise noted, all reactions were performed with 1 (0.15 mmol), 2 (0.10 mmol), L2 (10 mol%), CuBr2 (10 mol%), EtOH (1.0 mL), N-Em (20 mol%). The reaction was carried out at 20 ℃ for 36 h. Isolated yield. a The reaction was carried out for 48 h. b The reaction was carried out for 42 h.

Scheme 3 Scope of substrates. Unless otherwise noted, all reactions were performed with 1 (0.15 mmol), 3 (0.10 mmol), L2 (10 mol%), CuBr2 (10 mol%), EtOH (1.0 mL), N-Em (20 mol%). The reaction was carried out at 15 ℃ for 24 h. Isolated yield. a The reaction was carried out at 25 ℃ for 36 h.

Scheme 4 (A, B) Control experiments were performed under standard condi-tions. (C) Gibbs energy profiles for the Cu-catalyzed direct alkenylation reaction of 1a with 2a. Free energies in solution (in kcal/mol) at the (U) B3LYP-D3(BJ)/6-311 + G(d, p)-SDD/SMD (EtOH)//(U) B3LYP-D3 (BJ)/6-31G (d)/SDD/SMD(EtOH) level are displayed. 3D structures were generated by CYL view, key bond distances shown in units of Å [39].

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
