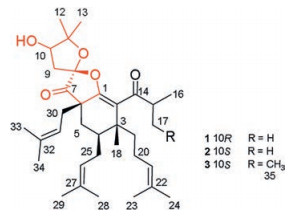
Figure 1.
Chemical structures of compounds 1–3.
Three new decarbonyl prenylphloroglucinols bearing unusual spirost subunits from Hypericum scabrum and their neuronal activities
Jie Ma , Guiyang Xia , Yingda Zang , Chuangjun Li , Jianbo Yang , Jiwu Huang , Jianjun Zhang , Yalun Su , Aiguo Wang , Dongming Zhang

Polycyclic polyprenyled acylphloroglucinols (PPAPs), with their fascinating biological profiles and intriguing complex molecular architectures, widely distributed in the family of Guttiferae (Clusiaceae), and have attracted considerable interest from both natural product and synthetic chemists over the past decades [1].Hypericum scabrum, belongs to the genus Hypericum of the Guttiferae family, exhibits various activities and has been used as an herbal treatment of hepatitis [2]. Previous chemical investigation on this plant revealed that a series of complex phloroglucinols have been isolated, which showed hepatoprotective and neuroprotective activity [3-11]. Inspired by the using St John′s (Hypericum perforatum) extracts to treat the moderate depression for many years and those neuroprotective phloroglucinols found, neuronal assays of components isolated from H. scabrum were undertaken. In our efforts to unearth the distinctive phloroglucinols from this title plant, three new PPAPs bearing unusual spirost subunits were isolated from the 95% EtOH extract of the aerial parts of H. scabrum. Compounds 1-3 (Fig. 1), a class of PPAPs, featured a new carbon skeleton with a rare 5, 5-spiroketal moiety. Herein, we describe the structural elucidation, postulated biosynthetic pathway, and neuron biological evaluation of those new compounds.
Compound 1 was isolated as colorless oil. [α]D20 + 37.9 (c 0.05, CH2Cl2). The molecular formula of C34H52O5 (found 540.3810 [M]+, calcd. for 540.3815) was deduced by its HREIMS data, corresponding to 9 degrees of unsaturation. The 1H NMR spectrum (CDCl3) of 1 showed three singlet methyls [δH 1.41 (H3-12), 1.29 (H3-13) and 1.17 (H3-18)], two characteristic doublet methyls of isopropyl [δH 1.14 (d, J = 6.9 Hz, H3-16), 1.12 (d, J = 5.4 Hz, H3-17)], six singlet isopentenyl methyls [δH 1.52 (H3-23), 1.62 (H3-24), 1.60 (H3-28), 1.69 (H3-29), 1.72 (H3-34), 1.57 (H3-33)]. The1H NMR spectrum also established three triplet olefinic protons resonanced at δH 4.93 (t, J = 7.5 Hz, H-21), 4.98 (t, J = 7.5 Hz, H-31), 5.05 (t, J = 6.6 Hz, H-26); a broad singlet methine at δH 4.05 (br s, H-10), and a septet methine (δH 3.33 (J = 6.3 Hz, H-15)). The 13C NMR and HSQC spectra of 1 classified those 34 carbons into 11 methyl (δC 17.7/C-23, 17.9/C-28, 18.1/C-33, 18.8/C-16, 19.9/C-17, 21.9/C-12, 22.5/C-18, 25.6/C-24, 25.88/C-29, 25.93/C-34, 26.8/C-13), 6 methylenes, 6 methines, and 11 quaternary carbons (2 ketone carbonyl carbons, 5 olefinic carbons included). The above data revealed that the structure of 1 should be a nor-phlorglucinols along with three isopentenyl moieties and an isobutyryl moiety. In addition, the aforementioned functionalities accounted for 6 of the 9 degrees of unsaturation, which implied the existence of three rings in the structure of 1.
Analysis of the HMBC spectrum gave rise to the conclusion that the architecture of 1 was mainly formed by two subunits, part A and B (Fig. 2a). The skeleton of part A (in black) of 1 closely resembled with (2R, 3R, 4S, 6R)-3-methyl-4, 6-di(3-methyl-2-butenyl)-2-(2-methyl-1-oxopropyl)-3-(4-methyl-3-pentenyl) cyclohexanone [11, 12]. The difference between them was mainly the presence of β-oxygenated α, β-unsaturated ketone functionality (-O-C1=C2-C14=O) in 1 instead of a 1, 3-diketone moiety, which was identified by the characteristic carbon chemical shifts of C-1 (δC 160.0), C-2 (δC 126.0) and C-14 (δC 207.1) [13]. The HMBC correlations from H3-18 to C-2, C-19, and C-4, from H2-19 to C-2, from H2-25 to C-5, and from H2-5 to C-30, C-7, and C-1 further established the presence of part A. Part B, a 2, 2-dimethyl-3-hydroxy-furan unit, was constructed by careful interpretation of the 1H-1H COSY correlation of H-9/H-10, as well as the well-resolved HMBC correlations from H2-9 to C-8, C-10 and C-11, from H-10 to C-8, and from H3-12/H3-13 to C-10/C-11, combing with the presence of two oxygen-bearing carbons C-10 (δC 77.1) and C-11 (δC 88.6). The clear HMBC cross-peaks from H2-9/H2-30/H2-5 to the ketone carbon C-7 (δC 208.0) deduced that C-7 was connected to C-6 and C-8. Finally, considering one remaining unsaturation, as well as the diagnostic ketal carbon C-8 (δC 109.6), the third circle of 1 was formed via an oxygen connected C-1 and C-8. Thus, the planar structure of 1 was constructed as shown in Fig. 1.
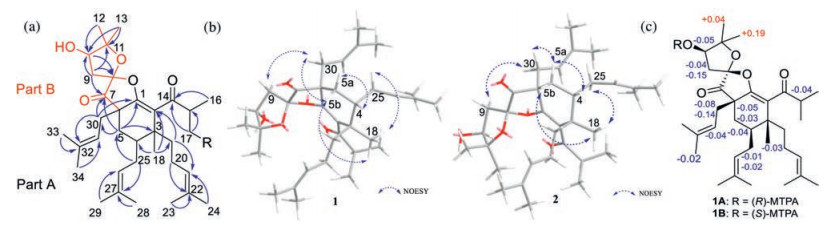


The relative configuration of 1 was confirmed on the basis of NOESY spectrum together with 3J coupling constants (Fig. 2b). In the 1H NMR spectrum, the large coupling constant (14.1 Hz) of H-5b/H-4 indicated their 1, 3-diaxial position in the six-membered ring with chair-like conformation [14]. In addition, the key NOESY correlations of H3-18/H-5b, H3-18/H2-25, and H-5b/H2-30 revealed that Me-18 and those two isopentenyl moieties attached to C-4/C-6 were in the same β-orientation. The NOESY cross-peaks of H-4/H-5a, H-4/H2-20, and H-4/H2-19 also indicated the H-4 and the isopentenyl linked to C-3 were α-orientated. The stereochemistry elucidation of spiro carbon C-8 was follows. Because the initial MM2-optimized structure for 1 revealed that those two five-membered rings divided by C-8 were almost perpendicular to each other, the NOESY correlations of H-9b/H-30a and H-9a/H-30b indicated that the C-9 attached at C-8 and the isopentyl moiety linked to C-6 were oriented on the same plane. However, special chemical atmosphere and the lack of NOESY correlations hampered the establishment of the relative configuration of C-10.
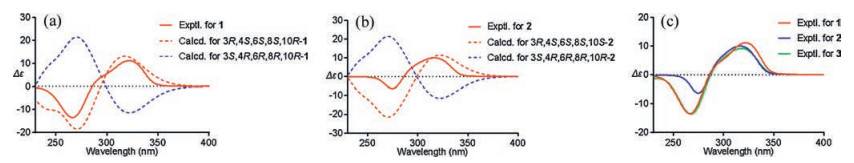
The absolute configuration of C-10 in 1 was validated through a modified Mosher′s experiment [15]. The prepared (S)- and (R)-MTPA esters of 1 were subjected to 1H NMR analysis, and the distinct values of the 1H NMR chemical shifts (Δδ = δS-MTPA-ester – δR-MTPA-ester) were summarized for the proton signals adjacent to C-10, as shown in Fig. 2c. According to these results, the absolute configuration of C-10 was confirmed to be R. To further support the configuration of the spiro-carbon C-8, a combination of computational NMR with DP4+ analysis and ECD methods were programmed. Calculated NMR with DP4+ analysis [16] was employed to establish the relative configuration of 1 from the two possibilities: 3R*, 4S*, 6S*, 8R*, 10R-1 (1a), 3R*, 4S*, 6S*, 8S*, 10R-1 (1b). Chemical shifts of isomers 1a and 1b were predicted using the gauge-independent atomic orbital (GIAO) method [17] with density functional theory (DFT) calculations in chloroform, using the polarizable continum model (PCM) model at the B3LYP/6-31+G (d, p) level. The experimental and calculated chemical shifts were statistically analyzed based on DP4+ probability. DP4+ analysis results showed that isomer 1b is the most reasonable configuration with a probability of 100% for the 13C data (Table S2 in Supporting information). A good linear correlation between the calculated 13C NMR chemical shifts and the experimental shifts was constructed (Fig. S29 in Supporting information). Afterwards, the time-dependent density functional theory (TD-DFT) ECD calculations were programmed for 3S, 4R, 6R, 8R, 10R-1 and 3R, 4S, 6S, 8S, 10R-1, respectively (Fig. 3a). The experimental ECD (Fig. 3a) of 1 well matched with the calculated ECD of 3R, 4S, 6S, 8S, 10R-1. Thus, the absolute structures of 1 was determined to be 3R, 4S, 6S, 8S, 10R-1.
Hyperscabin B (2), [α]D20 + 75.6 (c 0.18, CH2Cl2), displayed the same molecular formula of C34H52O5 as 1 based on the HREIMS data. The UV and IR spectral data of 2 were identical to those of 1, as shown in the Supporting information. Careful analysis of the NMR data, 2 closely resembled to those of 1 except for the very slight shifts in part B (1: δH 2.30, m, H-9a; 2.67, dd, J=14.5, 5.5 Hz, H-9b; 4.05, d, J = 5.7 Hz, H-10;2: δH 2.21, dd, J=15.0, 1.5 Hz, H-9a; 2.78, dd, J=15.0, 5.0 Hz, H-9b; 3.98, d, J = 4.8 Hz, H-10) (Table 1). It was estimated that stereogenic C-10 or C-8 might cause these differences.
 DownLoad:
CSV
DownLoad:
CSV

|
Like 1, the relative configurations of C-3, 4, 6, and 8 in 2 can be determined via the similar NOE correlations of H3-18/H2-30, H-5b/H2-25/H3-18 as 1, along with the NOE correlations of H2-9/H-30b (Fig. 2b). Due to the two perpendicular five-members rings attached to C-8, revealed by the initial optimized structure of 2, it indicated that the C-9 attached at C-8 and the isopentyl moiety linked to C-6 were in the same orientation. The configuration of C-10 was unconfirmed because of no relative NOESY signals and out of the amount. Thus, to further confirm the relative configuration elucidation of C-8 and C-10, the systematical 13C NMR calculation with DP4+ analysis was carried out for all four C-8 and C-10 relative possibilities: 3R*, 4S*, 6S*, 8S*, 10R*-2 (2a, isomer 1) and 3R*, 4S*, 6S*, 8S*, 10S*-2 (2b, isomer 2), 3R*, 4S*, 6S*, 8R*, 10S*-2 (2c, isomer 3) and 3R*, 4S*, 6S*, 8R*, 10R*-2 (2d, isomer 4). The results showed that 2b is the most reasonable configuration with a probability of 90.84% (Table S4 in Supporting information), which was further confirmed by the good linear correlation between the scaled calculated and experimental 13C NMR chemical shifts (Fig. S29 in Supporting information). The experimental ECD spectrum also corresponded well to the calculated spectrum for 3R, 4S, 6S, 8S, 10S-2 (Fig. 3b). So, the absolute configuration of 2 was finally established.
Compound 3, a colorless oil with [α]D20 +66.2 (c 0.08, CH2Cl2). Its HREIMS data showed a molecular ion peak at m/z 554.3936 [M]+, corresponding to a molecular formula of C35H54O5 (calcd. for 554.3971). There were 14 mass units more than those of 1 and 2. In addition, comparison of its NMR data (Table 1) with those of 2 indicated that the only difference between them was that the methyl group at C-17 in 2 was replaced by an ethyl group [CH2-17 (δH 1.95 m, 1.25 m; δC 26.3) and CH3-35 (δH 0.89 (t, 9.5); δC 11.9)] in 3. The parallel CD patterns of 3 and 2 suggested they had the same absolute configurations except of the ambiguous configuration at C-15 (Fig. 3c). Therefore, 3 was established as Hyperscabin C.
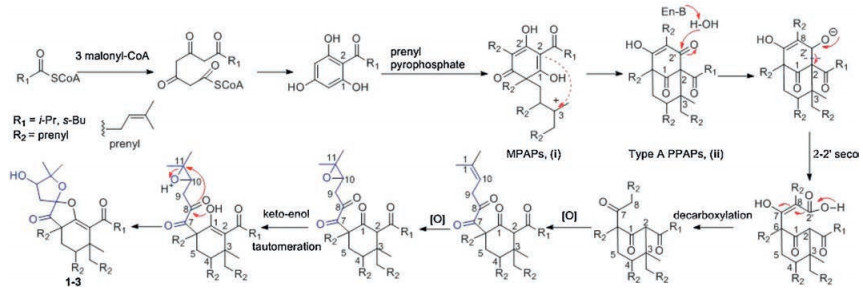
Biogenetically, the discovered PPAPs, isolated from H. scabrum form a unique family of structurally related spirost metabolites, are presumably derived from a common biosynthetic pathway. The polyketide-originated monocyclic polyprenylated acylphloroglucinols (MPAPs, ⅰ) were generated by a series of prenyl pyrophosphate procedures [1]. The intermediate ⅱ, possessing the bicyclo [3.3.1] nonane-2, 4, 9 trione core, was formed from the cyclization reactions of ⅰ [18]. Hyperscabrins A-C (1−3) could be considered as the PPAPs with the loss of C-2′ carbonyl in the phloroglucinol ring. The plausible biogenetic pathway was also proposed to be generated from ⅱ through the decarboxylation [19], followed by epioxidation, oxidation, keto-enol tautomeration and intramolecular cyclization reactions successively [20] (Scheme 1).
The isolated compounds were evaluated for their neuronal bioactivity. Compound 1 significantly increased cell viability (from 40.4% to 61.0%) measured in the model of oxygen and glucose deprivation/deoxygenation [21], which were more powerful than positive control drug potassium-(1-hydroxypentyl)-benzoate (PHPB, 47.4%). In addition, compounds 1 and 2 showed good inhibitory activity (81.8%, 83.2%) in 10 μmol/L against the reuptakion of [3H]-5-HT (Table S1 in Supporting information) [22].
In summary, three unprecedented prenylphloroglucinols (1–3) were isolated and elucidated via varies of chromatographic and spectroscopic methods from Hypericum scabrum. In terms of structure, they all possess a rare core, 5, 5-spiroketal subunit bearing a 6-member cyclic segment. They differ in the configurations of those groups and the isoprenyl side. It was difficult to establish its absolute configurations because of the difficulty to obtain crystals and their oily states of matter. Even so, we finally worked it out after attempts to use the NMR and ECD calculations. Their plausive biosynthetic pathways were rationalized in this article. Compounds 1 and 2 showed good inhibitory activity (81.8%, 83.2%) in 10 μmol/L against the reuptakion of [3H]-5-HT. Additionally, compound 1 significantly increased cell viability measured in the model of oxygen and glucose deprivation/deoxygenation. All in all, those compounds had potential neuron-protective and regulating activity, it was worthy of further investigation.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the Beijing Natural Science Foundation (No. 7194299), Fundamental Research Funds for the Central Universities (No. 3332018089), the CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-1-010), the Drug Innovation Major Project (No. 2018ZX09711001-002-010, 2018ZX09735006).
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.07.037.
X.W. Yang, R. Grossman, G. Xu, Chem. Rev. 118(2018) 3508-3558. doi: 10.1021/acs.chemrev.7b00551
X. Xu, B. Huangerhan, Medicinal Plants of Kazak, The Ethnic Publishing House, Beijing, 2008, pp. 57-59.
R.D. Liu, Y.L. Su, J.B. Yang, A.G. Wang, Phytochemistry 142(2017) 38-50. doi: 10.1016/j.phytochem.2017.06.011
J.W. Hu, W. Gao, F. Xu, et al., Biorg. Med. Chem. Lett. 27(2017) 4932-4936. doi: 10.1016/j.bmcl.2017.09.001
J.B. Yang, R.D. Liu, J. Ren, et al., J. Asian Nat. Prod. Res. 18(2016) 436-442. doi: 10.1080/10286020.2015.1123693
W. Gao, J.W. Hu, W.Z. Hou, et al., Tetrahedron Lett. 57(2016) 2244-2248. doi: 10.1016/j.tetlet.2016.04.026
W. Gao, W.Z. Hou, J. Zhao, et al., J. Nat. Prod. 79(2016) 1538-1547. doi: 10.1021/acs.jnatprod.5b01063
W. Gao, J.W. Hu, F. Xu, et al., Fitoterapia 115(2016) 128-134. doi: 10.1016/j.fitote.2016.10.003
R.D. Liu, J.B. Yang, J. Ma, et al., J. Asian Nat. Prod. Res. 16(2014) 717-723. doi: 10.1080/10286020.2014.917083
S.F. Nabavi, S.M. Nabavi, A.H. Moghaddam, et al., Toxicol. Environ. Chem. Rev. 94(2012) 779-785. doi: 10.1080/02772248.2012.671329
J. Ma, T.F. Ji, J.B. Yang, A.G. Wang, Y.L. Su, J. Asian Nat. Prod. Res. 14(2012) 508-514. doi: 10.1080/10286020.2012.680445
M.D. Shan, L.H. Hu, Z.L. Chen, J. Nat. Prod. 64(2001) 127-130. doi: 10.1021/np000362k
W.S. Li, A. Ma'ndi, J.J. Liu, et al., J. Org. Chem. 84(2019) 2596-2606. doi: 10.1021/acs.joc.8b03037
A.L. Piccinelli, O. Cuesta-Rubio, M.B. Chica, et al., Tetrahedron 61(2005) 8206-8211. doi: 10.1016/j.tet.2005.06.030
I. Ohtani, T. Kusumi, Y. Kashman, H. Kakisawa, J. Am. Chem. Soc. 113(1991) 4092-4096. doi: 10.1021/ja00011a006
N. Grimblat, M.M. Zanardi, A.M.J. Sarotti, J. Org. Chem. 80(2015) 12526-12534. doi: 10.1021/acs.joc.5b02396
M.W. Lodewyk, M.R. Siebert, D.J. Tantillo, Chem. Rev. 112(2012) 1839-1862. doi: 10.1021/cr200106v
X.W. Yang, M.M. Li, X. Liu, et al., J. Nat. Prod. 78(2015) 885-895. doi: 10.1021/acs.jnatprod.5b00057
W.J. Tian, Y. Yu, X.J. Yao, et al., Org. Lett. 16(2014) 3448-3451. doi: 10.1021/ol501333k
L.Z. Hu, H.C. Zhu, L. Li, et al., Sci. Rep. 6(2016) 27588. doi: 10.1038/srep27588
Y.D. Zang, X.Y. Song, C.J. Li, et al., Eur. J. Med. Chem. 143(2018) 438-448. doi: 10.1016/j.ejmech.2017.11.084
I. Artaiz, A. Zazpe, A. Innerarity, et al., Psychopharmacology 182(2005) 400-413. doi: 10.1007/s00213-005-0087-3

Figure 2
(a) Key HMBC
Table 1. 1H (400 MHz) and 13C NMR (100 MHz) data of compounds 1-3 in CDCl3 (25 ℃, J in Hz, δ in ppm).a
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


