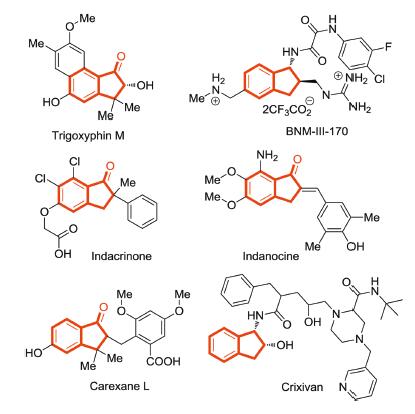
Figure 1.
Selected natural products and pharmaceuticals containing indanone cores.
Organophosphine bearing multiple hydrogen-bond donors for asymmetric Michael addition reaction of 1-oxoindane-2-carboxylic acid ester via dual-reagent catalysis
Haoran Hong , Hongyu Wang , Changwu Zheng , Gang Zhao , Yongjia Shang

Enantioselective nucleophilic phosphine catalysis [1-14] presents a powerful method for the construction of chiral functional molecules, and great advances have been made in particular for the annulations [15-33]. Many pharmaceuticals and natural products have been efficiently synthesized via this method [34-42]. However, little progress has been made on the research of new catalytic modes, which limited its applications in more powerful organic reactions. To expand the scope of asymmetric organophosphine catalysis, a new catalytic mode aroused by dual reagent, organophosphine and activated alkene, developed by our group [43] has been successfully applied in Mannich-type [44-48] and Strecker reactions [49], both of which are limited to the carbon-carbon bond formation with imines.
Indanones are very useful molecules which can be used as valuable synthons and pharmacophores ubiquitously found in numerous natural products [50-60] (Fig. 1). The synthesis of chiral indanone derivatives has received considerable attention of chemists. As our ongoing work on extending the applications of asymmetric dual-reagent catalysis for the construction of C-C bonds, we are interested in the Michael addition reaction [61-66] of indanone ester and activated olefin which can provide functionalized indanones with chiral quaternary carbons. Current asymmetric dual-reagent catalysis has been successfully applied in the Mannich-type reaction, Michael reaction and Strecker reaction with the organophosphine catalysts bearing double H-bonds (Fig. 2a). We envisioned that the interaction of the double H-bonds with the imines in addition to the carbonyl group affords an inflexible intermediate which is responsible for the high enantioselectivity.
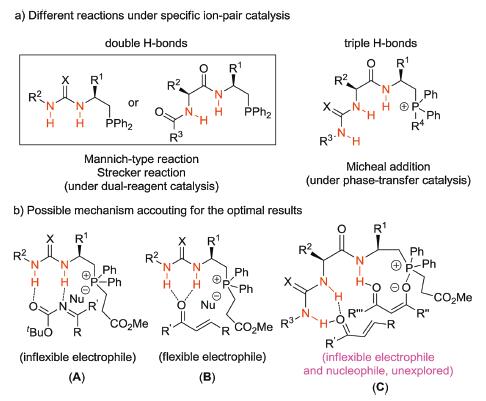
When this catalytic system is applied to the Michael reaction of α, β-unsaturated ketones, the double H-bonds will bind to the carbonyl group which results in a flexible intermediate and poor enantioselectivity might be observed (Fig. 2b). To further improve the enantioselectivity of Michael reaction of α, β-unsaturated ketones, rational design of new chiral quaternary phosphonium salts with triple H-bonds was introduced previously by our group (Fig. 2a), which enhanced the catalytic activity for improving enantioselectivities and diastereoselectivities in Michael reactions. We anticipated that multiple hydrogen bonds could strongly control both of the substrates in the transition state [67, 68], so asymmetric Michael addition reactions would be efficiently realized with high ee values and high dr values (Fig. 2b). Herein, asymmetric Michael addition reaction of indanone ester to the β-ester enones catalyzed by multiple hydrogen-binding organophosphine dual-reagents is investigated.
We initiated our study by selecting indene ketone esters 1 and β-ester enone 2 as the model substrates (Table 1). Inspired by our previous work, When the reaction was carried out in CH2Cl2 at room temperature with 5mol% of the organophosphines A-D (Fig. 3) and 5mol% of methyl acrylate (entries 1–4) [49], the desired product 3 was observed in good yield, but with poor enantioselectivity and diastereoselectivity. Hence, development of other powerful organophosphines to control the stereoselectivity for this reaction is demanded. A series of organophosphine catalysts E–J (Fig. 3) with multiple H-bonds featuring a dipeptide core structure were synthesized and used in model reaction. The first evaluation of catalysts E–H (Fig. 3) indicated that when R is the phenyl group, higher enantioselectivity and diastereoselectivity were observed (entries 5–8). Further investigation of the other amino acid skeleton revealed that organophosphine J was the optimum catalyst for the reaction (85% ee, 80:20 dr) (entries 9 and 10). In addition, when the ethyl group in 1 is changed into the tert-butyl group, the diastereoselectivity of the target product was improved (entry 11).
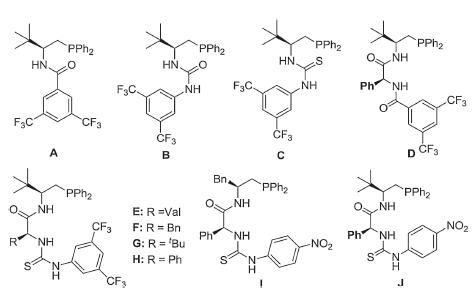
Following screening of several different solvents revealed that dichloromethane was more suitable for the reaction, with 84% ee and 90:10 dr achieved (for more details, see Supporting information). Various reaction temperatures were then investigated and it was found that the reaction was more selective at 0 ℃ (91% ee, 96:4 d.r.) (entries 11–13). Additionally, decreasing the catalyst loading led to reducing of yield and poor diastereoselectivity (entries 14 and 15). The reaction cannot proceed normally without adding methyl acrylate (entry 16). Herein we got the optimized reaction conditions as demonstrated with 5mol% of catalyst J and CH2Cl2 as a solvent at 0 ℃ (entry 12).
After optimized reaction conditions were found, we next turned our attention to exploring the indene esters (1b) and various activated olefins (2) in this Michael addition reaction. As summarized in Scheme 1, when R1 is fixed to ethyl group, both electronic nature and substituent effect on aromatic rings (when R2 = Ar) have little influence on the results. The corresponding products were obtained with uniformly excellent yields (89%–99%) and enantioselectivities (90%–95%) (3a-3f, 3h-3k). It is noteworthy that the enantioselectivity is slightly reduced for the enone bearing a strong electron-withdrawing group on aromatic ring (3g). Enones involving heteroaromatic or condensed ring also proved to be applicable (3l and 3m). Next, we studied the effects of different esters on the reaction products (such as R1 = Bn or tBu). When R1 becomes to be tert-butyl ester, reaction time is prolonged, yield and stereoselectivity are a little decreased (3n). However, when R1 becomes to be benzyl group, the reaction results are excellent with high yield and stereoselectivity achieved (3o). To our delight, the good enantioselectivity could be obtained at lower temperature when the terminal alkene was used (3q). The aliphatic (E)-ethyl-4-oxopent-2-enoate also worked in the reaction with good diastereoselectivity although with poor enantioselectivity (3p). We tried to replace compound 2 with chalcones, however, their activity was too low to initiate the reaction. We also tried α-tetralone derivatives be applied in this reaction instead of the indanone derivatives, but under the same optimized conditions, the expected product was not observed, inferred that the zwitterion was generated upon the mixing of organophosphine and methyl acrylate as a Brønsted base are too weak to not initiate the reaction.
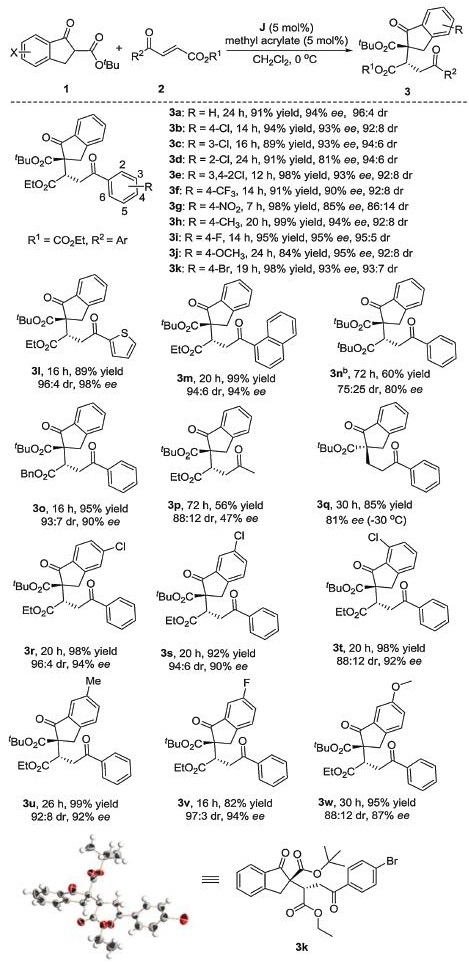
Next, we fixed 2a to evaluate the effect of different substituted substrates 1 on the reaction. The reaction demonstrated a broad substrate scope to provide the corresponding products in high yields and excellent stereoselectivity regardless of the electronic nature and the steric effect of substituents on the aryl ring of indanone esters substrates (3r-3w). Product 3k was dissolved with mixed solvent (hexane/CH2Cl2=50:1), volatilized at room temperature until the crystal particles can be obtained (more detail can see Supporting information). The absolute configuration of 3k was determined to be S, S by X-ray crystallographic analysis (CCDC No. 1869589)
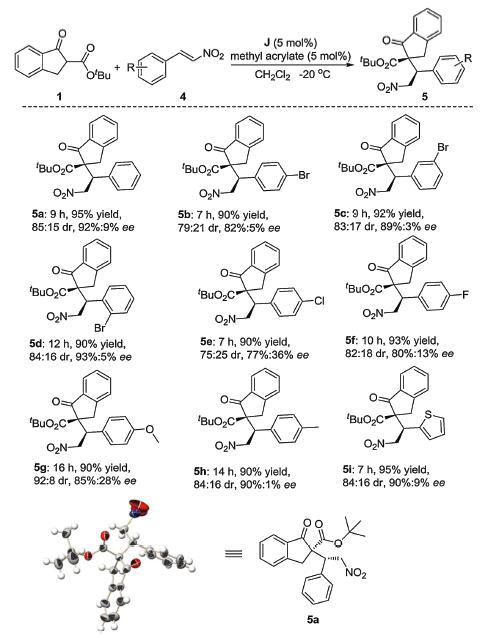
Among the Michael acceptors, nitroalkenes are very attractive as they can serve as masked functionality to be further transformed after the addition. The transformation such as the Nef reaction [69], the nucleophilic displacement [70], the reduction to an amino group [71-74] and the Meyer reaction [75] are only some examples that nitro groups can undergo. In recent years, a number of new efficient synthetic methods have been developed [76-80] on the application of nitroalkenes as Michael acceptors. After screening the reaction conditions with nitroalkenes (more detail can see Supporting information), the scope of the reaction was next investigated. As shown in Scheme 2, different substituents on the nitroalkenes were well tolerated. High yields and excellent enantioselectivities were obtained. These good results illustrated that the cooperation of multiple-hydrogen-bonds with dual-reagent ion pair catalyst based on the organophosphine will be a powerful method for improving enantioselectivities in organic synthesis. Product 5a was dissolved with mixed solvent (hexane/CH3OH=50:1), volatilized at room temperature until the crystal particles can be obtained (more detail can see Supporting information). The absolute configuration of 5a was determined to be S, R by X-ray crystallographic analysis (CCDC No. 1869591)
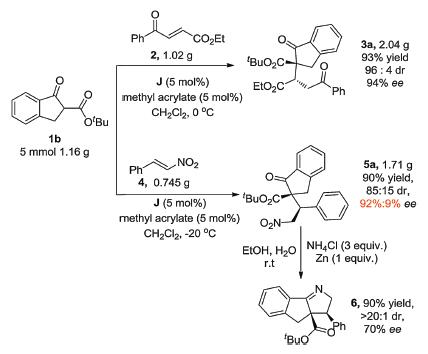
To demonstrate the synthetic utility of the products, we conducted gram-scale reaction to furnish 3a with 2.04g and in 93% yield upon isolation and 94% ee. At the same time, we also carried out gram-scale experiments on 5a to furnish 1.71g of the desired product in 90% yield upon isolation and 92% ee under the optimal reaction conditions. Furthermore, compound 5a was converted to phenyl dihydroindeno pyrrole carboxylate 6 easily under the reduction of Zn power in NH4Cl (aq.) and ethyl alcohol (Scheme 3).
To gain some insights into the mechanism of the reaction, some control experiments were conducted as shown in Fig. 3. Both yields and enantioselectivities decreased dramatically when N-methyl protected dipeptide-derived phosphonium K was employed (Fig. 4a). The results showed that hydrogen bonding of the catalyst was indispensable for the reactivity and enantioselectivity of reaction.
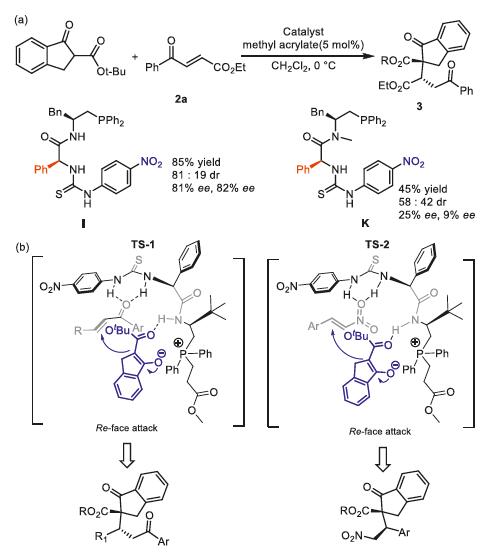
Based on these results, a plausible transition state TS-1 and TS-2 were proposed to explain the absolute configuration of the adducts (Fig. 4b). The zwitterion was generated upon the mixing of organophosphine and methyl acrylate, which then behaved as a mild Brønsted base to activate indanone esters. We proposed that the thiourea moiety of catalyst J might activate the electrophilic activated olefin via H-bonding interactions while the phosphonium ion pair and the amide of the catalyst J might direct the indanone esters through static electronic interaction and H-bonding interaction, respectively, to attack from the Re face to avoid steric repulsion from the phenyl and tertiary butyl groups of the catalyst [68].
In summary, we have developed an asymmetric dual-reagent catalyst system using amino-acid-derived dipeptides bearing multiple hydrogen bonds. This novel catalysis has demonstrated high catalytic efficiency in Michael additions to activated olefin with indanone esters, thus affording the corresponding indanone derivatives in excellent yields and enantioselectivity. Efforts toward a deeper understanding of the reaction mechanism and the application of catalytic model to other asymmetric transformations are in progress.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from Chinese Academy of Sciences (No. XDB 20020100) and the National Natural Science Foundation of China (Nos. 21572247, 21871282) is gratefully acknowledged.
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.07.026.
Z. Chen, G. Zhu, X. Zhang, et al., J. Org. Chem. 63 (1998) 5631-5635. doi: 10.1021/jo9721756
J.L. Methot, W.R. Roush, Adv. Synth. Catal. 346 (2004) 1035-1050. doi: 10.1002/adsc.200404087
Y. Chung, G.C. Fu, Angew. Chem. Int. Ed. 48 (2009) 2225-2227. doi: 10.1002/anie.200805377
B.J. Cowen, S.J. Miller, Chem. Soc. Rev. 38 (2009) 3102-3116. doi: 10.1039/b816700c
S.W. Smith, G.C. Fu, J. Am. Chem. Soc. 131 (2009) 14231-14233. doi: 10.1021/ja9061823
R. Sinisi, J. Sun, G.C. Fu, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 20652-20654. doi: 10.1073/pnas.1003597107
X.Y. Guan, Y. Wei, M. Shi, Eur. J. Org. Chem. (2011) 2673-2677.
H. Xiao, Z. Chai, G. Zhao, et al., Chem. Eur. J. 17 (2011) 10562-10565. doi: 10.1002/chem.201100850
Y.C. Fan, O. Kwon, Chem. Commun. 49 (2013) 11588-11619. doi: 10.1039/c3cc47368f
Y. Wei, M. Shi, Chem. Asian J. 9 (2014) 2720-2734. doi: 10.1002/asia.201402109
Y.L. Sun, Y. Wei, M. Shi, ChemCatChem 9 (2017) 718-727. doi: 10.1002/cctc.201601144
H. Guo, Y.C. Fan, Z. Sun, Y. Wu, O. Kwon, Chem. Rev. 118 (2018) 10049-10293. doi: 10.1021/acs.chemrev.8b00081
H. Ni, W.L. Chan, Y. Lu, Chem. Rev. 118 (2018) 9344-9411. doi: 10.1021/acs.chemrev.8b00261
H. Qiu, X. Chen, J. Zhang, Chem. Sci. 10 (2019) 10510-10515. doi: 10.1039/C9SC04073K
C. Zhang, X. Lu, J. Org. Chem. 60 (1995) 2906-2908. doi: 10.1021/jo00114a048
G. Zhu, Z. Chen, X. Zhang, et al., J. Am. Chem. Soc. 119 (1997) 3836-3837. doi: 10.1021/ja9644687
J.E. Wilson, G.C. Fu, Angew. Chem. Int. Ed. 45 (2006) 1426-1429. doi: 10.1002/anie.200503312
B.J. Cowen, S.J. Miller, J. Am. Chem. Soc. 129 (2007) 10988-10989. doi: 10.1021/ja0734243
A. Voituriez, A. Panossian, A. Marinetti, et al., J. Am. Chem. Soc. 130 (2008) 14030-14031. doi: 10.1021/ja806060a
H. Guo, Q. Xu, O. Kwon, J. Am. Chem. Soc. 131 (2009) 6318-6319. doi: 10.1021/ja8097349
X. Han, Y. Wang, F. Zhong, Y. Lu, J. Am. Chem. Soc. 133 (2011) 1726-1729. doi: 10.1021/ja1106282
Y. Fujiwara, G.C. Fu, J. Am. Chem. Soc. 133 (2011) 12293-12297. doi: 10.1021/ja2049012
Z. Xu, X. Lu, Tetrahedron Lett. 38 (1997) 3461-3464. doi: 10.1016/S0040-4039(97)00656-4
L. Jean, A. Marinetti, Tetrahedron Lett. 47 (2006) 2141-2145. doi: 10.1016/j.tetlet.2006.01.122
Y.Q. Fang, E.N. Jacobsen, J. Am. Chem. Soc. 130 (2008) 5660-5661. doi: 10.1021/ja801344w
X.F. Zhu, J. Lan, O. Kwon, J. Am. Chem. Soc. 125 (2003) 4716-4717. doi: 10.1021/ja0344009
R.P. Wurz, G.C. Fu, J. Am. Chem. Soc. 127 (2005) 12234-12235. doi: 10.1021/ja053277d
Y.S. Tran, O. Kwon, J. Am. Chem. Soc. 129 (2007) 12632-12633. doi: 10.1021/ja0752181
F. Zhong, X. Han, Y. Wang, Y. Lu, Chem. Sci. 3 (2012) 1231-1234. doi: 10.1039/c2sc00963c
Q. Zhang, L. Yang, X. Tong, J. Am. Chem. Soc. 132 (2010) 2550-2551. doi: 10.1021/ja100432m
S. Kramer, G.C. Fu, J. Am. Chem. Soc. 137 (2015) 3803-3806. doi: 10.1021/jacs.5b01944
W. Yao, X. Dou, Y. Lu, J. Am. Chem. Soc. 137 (2015) 54-57. doi: 10.1021/ja5109358
Y. Zhang, X. Tong, Org. Lett. 19 (2017) 5462-5465. doi: 10.1021/acs.orglett.7b02787
X. Han, F. Zhong, Y. Wang, Y. Lu, Angew. Chem. Int. Ed. 51 (2012) 767-770. doi: 10.1002/anie.201106672
K. Albertshofer, B. Tan, C.F. Barbas III., Org. Lett. 15 (2013) 2958-2961. doi: 10.1021/ol401087a
R.J. Lundgren, A. Wilsily, G.C. Fu, et al., Angew. Chem. Int. Ed. 52 (2013) 2525-2528. doi: 10.1002/anie.201208957
F. Zhong, X. Dou, Y. Lu, et al., Angew. Chem. Int. Ed. 52 (2013) 943-947. doi: 10.1002/anie.201208285
J.P. Michael, Nat. Prod. Rep. 21 (2004) 625-649. doi: 10.1039/b310689f
A.R. Pinder, Nat. Prod. Rep. 9 (1992) 491-504. doi: 10.1039/np9920900491
R.A. Jones, M.J. Krische, Org. Lett. 11 (2009) 1849-1851. doi: 10.1021/ol900360h
M.E. Abbasov, D. Romo, Nat. Prod. Rep. 31 (2014) 1318-1327. doi: 10.1039/C4NP00025K
D.H.A. Rocha, D.C.G.A. Pinto, A.M.S. Silva, Eur. J. Org. Chem. (2018) 2443-2457.
H.Y. Wang, K. Zhang, G. Zhao, et al., Angew. Chem. Int. Ed. 54 (2015) 1775-1779. doi: 10.1002/anie.201409342
Y.P. Lou, C.W. Zheng, R.M. Pan, et al., Org. Lett. 17 (2015) 688-691. doi: 10.1021/ol503712m
Q.W. Jin, C.W. Zheng, G. Zhao, G. Zou, Tetrahedron 74 (2018) 4134-4144. doi: 10.1016/j.tet.2018.06.030
X. Ji, W.G. Cao, G. Zhao, Tetrahedron 73 (2017) 5983-5992. doi: 10.1016/j.tet.2017.08.031
L.J. Xu, H.Y. Wang, C.W. Zheng, G. Zhao, Adv. Synth. Catal. 359 (2017) 2942-2948. doi: 10.1002/adsc.201700321
R.M. Pan, W.G. Cao, G. Zhao, et al., Tetrahedron 73 (2017) 2349-2358. doi: 10.1016/j.tet.2017.03.005
H.Y. Wang, C.W. Zheng, Z. Chai, J.X. Zhang, G. Zhao, Nat. Commun. 7 (2016) 12720-12728. doi: 10.1038/ncomms12720
K. Kundu, J.V. McCullagh, A.T. Morehead, J. Am. Chem. Soc. 127 (2005) 16042-16043. doi: 10.1021/ja0564416
R.S. Senaiar, J.A. Teske, D.D. Young, A. Deiters, J. Org. Chem. 72 (2007) 7801-7084. doi: 10.1021/jo7013565
M.A. Ernst-Russell, C.L.L. Chai, J.H. Wardlaw, J.A. Elix, J. Nat. Prod. 63 (2000) 129-131. doi: 10.1021/np9903245
X. Zhang, J.K. Xu, J. Wang, et al., J. Nat. Prod. 70 (2007) 24-28. doi: 10.1021/np060449r
C. Fan, W. Wang, Y. Wang, G. Qin, W. Zhao, Phytochemistry 57 (2001) 1255-1258. doi: 10.1016/S0031-9422(01)00168-6
H.O. Saxena, U. Faridi, A.S. Negi, et al., Bioorg. Med. Chem. Lett. 18 (2008) 3914-3918. doi: 10.1016/j.bmcl.2008.06.039
R. Sheng, Y. Xu, Y. Hu, et al., Eur. J. Med. Chem. 44 (2009) 7-17. doi: 10.1016/j.ejmech.2008.03.003
T.L. Fu, I.J. Wang, Dyes Pigm. 76 (2008) 158-164. doi: 10.1016/j.dyepig.2006.07.030
T.L. Fu, I.J. Wang, Dyes Pigm. 76 (2008) 590-595. doi: 10.1016/j.dyepig.2007.02.007
X. Li, J. Han, J. Meng, et al., J. Mol. Struct. 874 (2008) 28-33. doi: 10.1016/j.molstruc.2007.03.028
J. Han, J. Meng, J. Photochem, Photobiol. C: Photochem. Rev. 10 (2009) 141-147. doi: 10.1016/j.jphotochemrev.2009.10.001
S. Shirakawa, A. Kasai, T. Tokuda, K. Maruoka, Chem. Sci. 4 (2013) 2248-2252. doi: 10.1039/c3sc22130j
A.E. Sheshenev, E.V. Boltukhina, A.J.P. White, K.K. Hii, Angew. Chem. Int. Ed. 52 (2013) 6988-6991. doi: 10.1002/anie.201300614
F. Zhong, X. Dou, Y. Lu, et al., Angew. Chem. Int. Ed. 52 (2013) 943-947. doi: 10.1002/anie.201208285
X.Y. Wu, W.G. Cao, G. Zhao, et al., Adv. Synth. Catal. 355 (2013) 2701-2706. doi: 10.1002/adsc.201300383
B. Huang, C. Li, J. Zhang, et al., Org. Lett. 19 (2017) 5102-5105. doi: 10.1021/acs.orglett.7b02365
(a) F. Yin, A. Garifullina, F. Tanaka, Org. Biomol. Chem. 15 (2017) 6089-6092;
(b) S.X. Cao, J.X. Wang, Z.J. He, Chin. Chem. Lett. 29 (2018) 201-204;
(c) K. Yang, Z.Y. Ma, H.X. Tong, X.Q. Sun, Z.Y. Li, Chin. Chem. Lett. 31 (2020) 3259-3262.
(a) D.D. Cao, J.X. Zhang, H.Y. Wang, G. Zhao, Chem. Eur. J. 21 (2015) 9998-10002;
(b) D.D. Cao, G.S. Fang, G. Zhao, et al., J. Org. Chem. 81 (2016) 9973-9982.
(a) C.J. Wang, Z.H. Zhang, X.Q. Dong, X.J. Wu, Chem. Commun. (2008) 1431-1433;
(b) X. Fang, C.J. Wang, Chem. Commun. 51 (2015) 1185-1197.
J.U. Nef, Justus Liebigs Ann. Chem. 284 (1894) 263-291.
R. Tamura, A. Kamimura, N. Ono, Synthesis (1991) 423-434.
A.K. Beck, D. Seebach, Chem. Ber. 124 (1991) 2897-2911. doi: 10.1002/cber.19911241233
M.A. Poupart, G. Fazal, S. Goulet, L.T. Mar, J. Org. Chem. 64 (1999) 1356-1361. doi: 10.1021/jo9815204
A.G.M. Barrett, C.D. Spilling, Tetrahedron Lett. 29 (1988) 5733-5734. doi: 10.1016/S0040-4039(00)82175-9
D.H. Loyd, D.E. Nichols, J. Org. Chem. 51 (1986) 4294-4298. doi: 10.1021/jo00372a037
M.J. Kamlet, L.A. Kaplan, J.C. Dacons, J. Org. Chem. 26 (1961) 4371-4375. doi: 10.1021/jo01069a046
A.G.M. Barrett, G.G. Graboski, Chem. Rev. 86 (1986) 751-762. doi: 10.1021/cr00075a002
R. Ballini, R. Castagnani, M. Petrini, J. Org. Chem. 57 (1992) 2160-2162. doi: 10.1021/jo00033a045
O.M. Berner, L. Tedeschi, D. Enders, Eur. J. Org. Chem. (2002) 1877-1894.
N.J.A. Martin, L. Ozores, B. List, J. Am. Chem. Soc. 129 (2007) 8976-8977. doi: 10.1021/ja074045c
N. Lv, Z. Chen, Y. Liu, Z. Liu, Y. Zhang, Adv. Synth. Catal. 361 (2019) 4140-4146. doi: 10.1002/adsc.201900427

Scheme 1 Substrate scope of indanone derivative. Reactions were carried out with 1 (0.1 mmol), 2 (0.1 mmol), and methyl acrylate (0.5% mol) in the presence of chiral phosphine J (0.5% mol) in DCM (1.0 mL) at 0 ℃.

Scheme 2 Substrate scope of nitroalkenes. Reactions were carried out with 1 (0.1 mmol), 4 (0.1 mmol), and methyl acrylate (0.5% mol) in the presence of chiral phosphine J (0.5% mol) in CH2Cl2 (1.0 mL) at -20 ℃.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



