Citation:
Zhang Shengrui, Wang Qin, Yang Jiajun, Yang Xiao-Feng, Li Zheng, Li Hua. A photocalibrated NO donor based on N-nitrosorhodamine 6G upon UV irradiation[J]. Chinese Chemical Letters,
2019, 30(2): 454-456.
doi:
10.1016/j.cclet.2018.03.011
A photocalibrated NO donor based on N-nitrosorhodamine 6G upon UV irradiation
English
A photocalibrated NO donor based on N-nitrosorhodamine 6G upon UV irradiation
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, China
b.
Shaanxi Key Laboratory of Catalysis, School of Chemistry and Environment Science, Shaanxi University of Technology, Hanzhong 723000, China
c.
College of Life Sciences, Northwest University, Xi'an 710069, China
d.
College of Chemistry and Chemical Engineering, Xi'an Shiyou University, Xi'an 710065, China
Received Date:
12 January 2018 Accepted Date:
06 March 2018 Revised Date:
05 March 2018 Available Online:
22 February 2019
Abstract:
A photocalibrated NO donor, N-nitrosated rhodamine 6G acid (NOG) was designed for monitoring the kinetics or the dose of NO release in a real-time fashion with spectroscopic methods Upon irradiation, a drastic fluorescence turn-on was observed, and the NO release from NOG was not interfered by biothiols. Furthermore, this NO donor enabled time- and site-controlled NO release in living cells. With these merits, NOG can be a useful molecular tool in NO biology.
As a reactive nitrogen species produced in living systems, nitric oxide (NO) plays important roles in the apoptosis, cardiovascular, immune, and central nervous systems [1-5]. The roles of NO in biological systems are related with the location, time, and dose, especially the complex microenvironments [6, 7]. Since the novel biological functions of NO, various NO donors employed as exogenous sources of NO have been constructed [8]. However, most of them release NO via spontaneous decomposition, which is practically impossible to liberate NO in a time- and site-controlled manner, and hard to discriminate their NO release from typical systemic donors such as glycerin trinitrate. Therefore, the construction of NO donors with a high spatiotemporal control and little interference was indispensable for investigating NO physiology and developing potential therapeutic agents.
Since photochemical reactions are easily controlled by the interval and position of light exposure, photosensitive NO donors using light as a localized stimulus for NO delivery have the ability to surmount the above difficulties [9]. Over the past few years, some photo-controllable NO donors have been reported, such as metal nitrosyls [10], NONOates [11], and so on. For example, Fry and Mascharak et al. designed and synthesized several metal nitrosyls (NO complexes of metals) that rapidly release NO when exposed to light of various wavelengths [12]. Miyata et al. reported a novel 2, 6-dimethylnitrobenzene based NO donor which can be controlled by the two-photon excitation technique [9]. Nakagawa et al. developed a yellowish-green-light-controllable NO donor and the NO release from it was detected by ESR spin trapping and a NO fluorescent probe [13]. Though straightforward, these methods have their limitations, such as biological background absorption and cell damage due to activating UV light, toxicity related to photoproducts (the remaining metal complexes), stability in biological media, use of UV–vis absorption spectroscopy or fluorescence turn-off mechanism.
To overcome these limitations, Yang et al. reported a series of N-nitrosated naphthalimides as novel NO donors whose NO release can be triggered by light, accompanying by a drastic fluorescence turn-on, which was propitious to monitor the kinetics or dose of NO release in vitro studies [14]. Subsquently, Tang et al. reported a ratiometric fluorogenic NO photoreleaser (CNNO) for imaging and tracking of NO release in live cells [15]. However, such NO donors are still very limited, much more efficient NO donors are looking forward to being developed.
In this work, we designed and synthesized a novel NO donor, N-nitrosated rhodamine 6G acid (NOG) using rhodamine 6G (R6G) as fluorophore, in which one molecular probe can release two molecules of NO. Interestingly, very recently, Yang et al. have developed such photocalibrated NO donors derivated from rhodamine dyes, and applied these donors in superresolution imaging of subcellular organelles, such as mitochondria and lysosomes [16, 17]. Our rationale is depicted in Scheme S1 (Supporting information). NO release from NOG may be triggered by UV irradiation at 365 nm and accompanied by a drastic fluorescence turn-on, which was convenient to quantitate NO dose in vitro. Importantly, such a photocalibrated NO donor is not interfered with biothiols. In addition, NOG exhibits the merits of spatiotemporal controlling NO release in living cells.
R6G acid has been extensively used as fluorophores because of their excellent photophysical properties [18-20]. More importantly, spirocyclic form of R6G is colorless and non-fluorescent, whereas addition of targeted moleculars leads to ring-opened form, accompanying by a drastic fluorescence emission and a color change from colorless to pink. On the other hand, electronwithdrawing substituted N-nitrosamines are well-known to yield NO upon photolysis [21-23]. Thus NOG was designed as a novel NO donor for NO release. Upon phototriggered decomposition, the byproduct, the anilinyl radical, is oxidative and expected to be readily reduced in situ to afford R6G acid.
Since compound R6G acid is the expected product of NOG upon phototriggered by UV light at 365 nm, we first examined the differences of the absorption and fluorescent spectra between NOG and R6G acid. As shown in Fig. S1 (Supporting information), their absorption and fluorescence spectra both showed significant differences. The NOG exhibited non-absorption in the range of 350 nm to 600 nm and non-fluorescent, which is attributed to the colorless and non-fluorescent spirocyclic form of R6G acid. However, R6G acid displayed absorption with a maximum at 520 nm, and showed an intense emission band with a maximum at 547 nm. Thus, this low background fluorescence of the donor itself and the strong fluorescence of the expected product should be highly desirable for monitoring the dose of NO release.
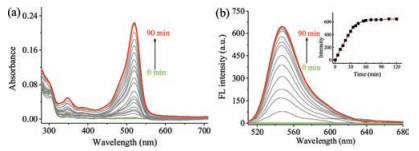
With NOG in hand, photoinduced NO release was firstly examined in aqueous phosphate buffer (20 mmol/L, pH 7.4) at room temperature. Distinct changes in the absorption and emission of NOG were observed after the solutions were irradiated by UV light at 365 nm. Upon irradiation, the absorption at 346 nm and 520 nm increased abruptly, and reached the maximum after ~60 min (Fig. 1a). Meanwhile, fluorescence emission spectroscopy of the solution was observed with excitation wavelength at 500 nm (Fig. 1b). The fluorescence intensity centered at 547 nm was also increased steadily and gradually leveled off, indicating the appearance of R6G acid. This experiment was in agreement with the proposed decomposition pathway of NOG (Fig. 2a). In addition, the drastic fluorescence turn-on which accompanies the NO release from NOG, facilitated to monitor the kinetics and dosage of NO generation in a real-time fashion. Accordingly, after irradiation with 365 nm light for 60 min, the maximum concentration of NO released from 2.0 mmol/L NOG was determined to be 3.96 mmol/L based on the fluorescence calibration curve established from the relationship between fluorescence intensity and concentration of R6G acid (Fig. S2 in Supporting information).
Figure 1
Figure 1.
Changes of the UV–vis absorption spectra (a) and fluorescence emission spectra (b) of NOG solution (2.0 μmol/L) upon UV-irradiation at 365 nm. The inset of (b) shows fluorescence emission intensity at 547nmwith respect to the duration of UV-irradiation.
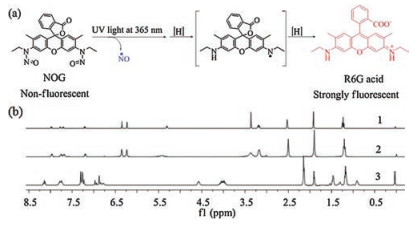
Figure 2.
(a) Pathway of NO release and fluorophore generation upon UV-triggered decomposition of NOG. (b) The 1H NMR spectra of R6G acid upon photolysis of NOG (1), R6G acid from independent synthesis (2), and NOG (3).
Then, MS and NMR analysis were further conducted to confirm that the NO release is indeed due to NOG upon UV-irradiation. An intense peak at m/z 415.2018 found in the MS spectrum further proved the formation of R6G acid (calcd. 415.2016 for C26H27N2O3+)
(Fig. S3 in Supporting information). Moreover, as shown in Fig. 2b, the 1H NMR spectrum of the isolated product is consistent with the structure of R6G acid. Generally speaking, this study has confirmed that the decomposition of NOG is in line with our proposed mechanism.
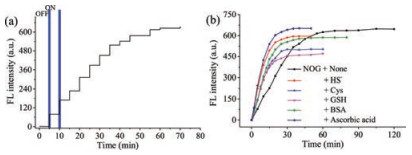
The photosensitivity and stability of NOG were investigated for several light on/off cycles. As shown in Fig. 3a, the fluorescence intensity remained unchanged without irradiation and enhanced only after irradiation at 365 nm, suggesting that NO release from NOG could be regulated with temporal precision. Additionally, the fluorescence intensity of NOG and R6G acid remained stable after irradiated by a xenon lamp within 30 min, indicating their predominant photostability (Fig. S4 in Supporting information).
Figure 3
Figure 3.
(a) Controlled release profile of 2 μmol/L NOG upon irradiation at 365 nm light for different on/off cycles, λex = 500 nm, λem = 547. (b) Time-dependent fluorescence enhancement (λex/λem = 500/547 nm) of a NOG solution (2.0 μmol/L) in phosphate buffer (20 mmol/L, pH 7.4) with 1% DMSO, upon continuous.
Subsquently, the influence of ascorbic acid and reactive sulfur species thiols on NO release from NOG was performed. As shown in Fig. 3b, when ascorbic acid is present, decomposition of NOG was completed in 30 min, which obviously show that the presence of ascorbic acid has dramatically shortened the duration of decomposition of NOG as well as reactive sulfur species thiols, such as HS¯, GSH, Cys and BSA. The phenomenon from the above experiment strongly demonstrates the nature medium would not affect the monitor of the dose of NO release from NOG.
According to the literature [24], transnitrosation may happen between a nitrosamine and a thiol, which would unexpectedly turn on a drastic fluorescence signal and not conducive to control the NO release. Therefore, the reactivity of NOG toward thiols was performed. The NOG was mixed with representative biothiols in neutral phosphate buffer and stirred in the dark for 24 h. As shown in Fig. S5 (Supporting information), NOG was totally not interfered with the potential nucleophilic attack from these biothiols.
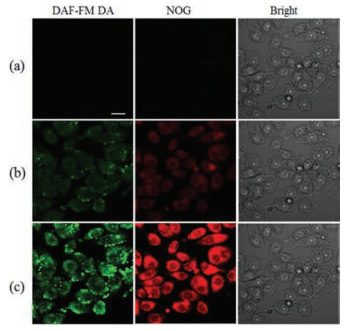
Encouraged by the above-mentioned results, the release of NOG in cellular studies was evaluated. Before cell imaging experiments, cytotoxicity of NOG was studied with the MTT assay. SMMC-7721 cells were incubated with up to 30 μmol/L of NOG for 24 h. Cell growth was not inhibited (Fig. S6 in Supporting information), indicating that NOG (up to 30 μmol/L) exhibits minimal toxic effects on cell viability. Subsquently, photoinduced NO release from NOG in SMMC-7721 cells was investigated with 3-amino, 4- aminomethyl-2', 7'-difluorescein, diacetate (DAF-FM DA), which can be hydrolyzed intracellularly to a fluorogenic NO probe, DAFFM [25]. SMMC-7721 cells were preincubated with NOG and DAFFM DA for 15 min, and imaged by confocal microscopy. Essentially, no fluorescence signal can be observed (Fig. 4a). Upon irradiation of the cells with light at 365 nm, green fluorescence emission from DAF-FM and yellow fluorescence emission from R6G acid were clearly captured. A significant time-dependent fluorescence enhancement was clearly observed (Figs. 4b and c). These results suggested that NOG enabled time-controlled NO release in living cells.
Figure 4
Figure 4.
Photocontrolled NO release in SMMC-7721 cells. Fluorescence image without photoirradition (a) and with photoirradiation at 365 nm for 2 min (b) and 10 min (c). Scale bar represents 20 μm.
To confirm that NO release could be confined to a specified area, SMMC-7721 cells were preincubated with NOG, and then the cells within a specified region were stimulated with an argon laser (488 nm) via a confocal fluorescence microscope. As shown in Fig. S7 (Supporting information), the fluorescence intensity was increased only within and around the irradiated area. Thus, NOG enabled site-controlled NO release in living cells.
In conclusion, we have constructed a novel photocalibrated NO donor, NOG. Upon UV irradiation, a drastic fluorescence turn-on accompanies the release of NO, which facilitated the quantification of NO release. Moreover, the decomposition pathway is not disturbed by nature of medium, and the biological thiols do not cause the NO release. We have also found that NOG enabled timeand site-controlled NO release in living cells. We expect this donor to receive wide applications.
Acknowledgment
This research was supported by the National Natural Science Foundation of China (Nos. 21475105, 21675123).
Figure 1
Changes of the UV–vis absorption spectra (a) and fluorescence emission spectra (b) of NOG solution (2.0 μmol/L) upon UV-irradiation at 365 nm. The inset of (b) shows fluorescence emission intensity at 547nmwith respect to the duration of UV-irradiation.
Figure 2
(a) Pathway of NO release and fluorophore generation upon UV-triggered decomposition of NOG. (b) The 1H NMR spectra of R6G acid upon photolysis of NOG (1), R6G acid from independent synthesis (2), and NOG (3).
Figure 3
(a) Controlled release profile of 2 μmol/L NOG upon irradiation at 365 nm light for different on/off cycles, λex = 500 nm, λem = 547. (b) Time-dependent fluorescence enhancement (λex/λem = 500/547 nm) of a NOG solution (2.0 μmol/L) in phosphate buffer (20 mmol/L, pH 7.4) with 1% DMSO, upon continuous.
Figure 4
Photocontrolled NO release in SMMC-7721 cells. Fluorescence image without photoirradition (a) and with photoirradiation at 365 nm for 2 min (b) and 10 min (c). Scale bar represents 20 μm.

 DownLoad:
DownLoad:

 下载:
下载:





