 图 3
催化剂前驱体FT-IR 谱图
Figure 3.
FT-IR profiles of the catalyst precursors
图 3
催化剂前驱体FT-IR 谱图
Figure 3.
FT-IR profiles of the catalyst precursors
引用本文:
陈俊军, 高文桂, 王华, 纳薇. CaO对Cu-ZnO-ZrO2催化CO2加H2合成甲醇性能影响[J]. 燃料化学学报,
2016, 44(4): 437-448.

Citation: CHEN Jun-jun, GAO Wen-gui, WANG Hua, NA Wei. Effect of CaO on the performance of Cu-ZnO-ZrO2 catalyst for methanol synthesis from CO2 and H2[J]. Journal of Fuel Chemistry and Technology, 2016, 44(4): 437-448.
Citation: CHEN Jun-jun, GAO Wen-gui, WANG Hua, NA Wei. Effect of CaO on the performance of Cu-ZnO-ZrO2 catalyst for methanol synthesis from CO2 and H2[J]. Journal of Fuel Chemistry and Technology, 2016, 44(4): 437-448.
CaO对Cu-ZnO-ZrO2催化CO2加H2合成甲醇性能影响
摘要:
用CaO作为改性助剂,采用并流共沉淀法制备了CuO:ZnO:ZrO2为5:4:1(物质的量比),CaO添加量为0、1%、2%、4%、8%、16%(摩尔分数)的六组催化剂。用X射线衍射(XRD)、微商热重(TG-DTG)、傅里叶红外(FT-IR)、N2吸附脱附(BET)、X射线光电子能谱(XPS)、氢气程序升温还原(H2-TPR)、CO2程序升温脱附(CO2-TPD)、NH3程序升温脱附(NH3-TPD)对催化剂进行了表征。用自制固定床评价了催化剂活性。结果表明,添加CaO后,催化剂路易斯酸性和表面碱性增强;催化剂母体中高温碳酸盐含量增加,热稳定性增强,CuO颗粒粒径变小,Cu-Zn协同作用增强,Cu比表面积增大,分散性变好。催化剂活性受到表面酸碱性、铜比表面积、Cu-Zn协同作用和铜分散性共同影响。当CaO为2%时,铜比表面积为79.3 m2/g、铜分散度为34.8%、CO2转化率为24.55%、甲醇选择性为19.01%、甲醇收率为0.044 g/(gcat·h),催化剂活性最好。过量CaO占据催化剂孔道和覆盖表面活性位,使催化剂路易斯酸性和表面碱性过强,CuO与H2有效接触减少,CO2难以脱附,催化活性下降。因此,适量CaO(2%)添加可促进CO2加氢反应合成甲醇。
-
关键词:
- 并流共沉淀法
- / CaO
- / Cu-ZnO-ZrO2
- / CO2加H2
- / 甲醇
English
Effect of CaO on the performance of Cu-ZnO-ZrO2 catalyst for methanol synthesis from CO2 and H2
Abstract:
CuO:ZnO:ZrO2=5:4:1 (molar ratio) catalysts were prepared with CaO doping of 0,1%, 2%, 4%, 8%, 16% (molar fraction) by cocurrent-flow co-precipitation. X-ray diffraction (XRD), thermal analysis(TG-DTG), Fourier infrared (FT-IR), N2 adsorption desorption (BET), X-ray photoelectron spectroscopy (XPS), hydrogen temperature programmed reduction (H2-TPR), CO2 temperature programmed desorption (CO2-TPD) and NH3 temperature programmed desorption (NH3-TPD) were used to characterize the catalysts. The catalyst activity was evaluated with a lab-made fixed bed reactor. Results show that CaO doping enhances Lewis acid and surface alkaline of the catalyst, increases the amount of high temperature carbonate in the catalysts, improves the thermal stability, reduces the CuO particle size, enhances the synergistic effect of Cu-Zn, increases the Cu specific surface area and the Cu dispersion. The catalyst activity is influenced by the surface acidity, the specific surface area of copper, the synergistic effect of Cu-Zn and the dispersion of copper. When the doping amount of CaO is 2%, the copper specific surface area is 79.3 m2/g, the dispersion degree of copper is 34.8%, the CO2 conversion is 19.01%, the selectivity of methanol is 24.55% and the yield of methanol is 0.044 g/(gcat·h), catalyst activity is the highest. With the amount of CaO increasing, the excessive CaO occupies the catalyst pore and covers the surface active sites, the Lewis acid and the surface alkaline of the catalysts become so strong that the effective contact of CuO and H2 is reduced, CO2 is difficult to desorb, resulting in decrease of catalytic activity. Therefore, the proper doping amount of CaO (2%) can promote the synthesis of methanol through CO2 hydrogenation.
-
Key words:
- cocurrent-flow co-precipitation
- / CaO
- / Cu-ZnO-ZrO2
- / CO2 and H2
- / methanol
-
温室气体的过多排放给人类带来巨大挑战,而CO2是造成全球气体变暖的主要温室气体。现在,全球平均气温与同期相比升高0.6-1℃[1, 2]。将空气中大量的CO2(750Gt[3])转变为有价值的化学品,既具有经济意义,又具有环保价值,是缓解全球变暖的有效途径,已引起研究者广大关注。
CO2合成甲醇是一种理想利用途径。甲醇毒性低,易储存运输,是一种有机液相储氢化合物,可以用作车用燃料。M85(85%(体积分数)甲醇和15%无铅汽油)已经应用于甲醇汽车,且不用对车进行大量技术改进。M85自从2008年进入中国至现在,已使用3.8×106m3甲醇作为车用燃料[4]。另外,甲醇还可用于燃料电池燃料和基础化学原料。由于CO2标准生成热为-393.54kJ/mol,具有很大的化学惰性[5],要将其转化为甲醇,催化剂的研发就成为关键和难点。
CO2加氢合成甲醇催化剂是在工业合成气制甲醇催化剂Cu-ZnO-Al2O3体系上发展起来的。CO2加氢合成甲醇产生水,Al2O3有强的亲水性,水会降低催化剂活性位的稳定性,因此,Cu-ZnO-Al2O3不适合CO2加氢合成甲醇。ZrO2在还原和氧化条件下都具有好的热稳定性[6],可促进铜氧化态的还原[7],Ma等[8]认为,Cu-ZnO-ZrO2催化剂可加强Cu-Zn间的相互作用,对CO2加氢合成甲醇具有较好活性。对于铜基催化剂,助剂主要可改变Cu的分散度、颗粒粒径、铜与载体的相互作用、催化剂的热稳定性、酸碱性,从而改变催化剂的催化活性。助剂主要包括IIIA元素、过渡金属元素、稀土元素、碱金属元素和碱土金属元素。Sloczynski等[9]发现,Cu-ZnO-ZrO2催化剂添加B、Ga、In后,CO2加氢合成甲醇反应活性有很大区别。添加Ga2O3,催化剂具有高的收率,加入In2O3,催化剂活性显著减低。Slozynski等报道[10],Cu-ZnO-ZrO2加入Mg、Mn助剂后,催化剂铜分散性增加,Mg、Mn助剂趋向催化剂表面聚集,催化活性遵循CuZnZr<CuZnZrMg<CuZnZrMn。Ban等[11]发现,La、Ce添加Cu-ZnO-ZrO2中,可促进CO2加氢合成甲醇的生成,Nd、Pr使得Cu-ZnO-ZrO2具有较低的催化活性。Tan等[12]发现,Cr/ZnO催化合成气制备异丁醇时,碱金属助剂强烈影响催化剂的还原性和关键组分的组成。Zhong等[13]报道,碱土金属氧化物具有很高热稳定性,可防止CuO-ZrO2催化剂烧结,增加催化剂中碱性位的数量和强度。
CaO为碱土金属氧化物,较活泼,对酸性CO2有较好吸附作用,可作为改性助剂。Elias等[14]发现,Ni-Al2O3体系加入CaO后,催化剂稳定性增加,表面酸性降低。CaO可吸附催化反应中产生的水,提高催化剂的热稳定性。Cu-ZnO-ZrO2为CO2加氢合成甲醇理想的催化体系,向其中加入CaO,以此来提高催化活性,目前报道极少。研究用CaO改性Cu-ZnO-ZrO2催化剂并用于CO2加氢合成甲醇,探究CaO对其表面结构、催化性能的影响。
1 实验部分
1.1 催化剂的制备
采用并流共沉淀法制备CaO改性Cu-ZnO-ZrO2催化剂,其中,CuO∶ZnO∶ZrO2为 5∶4∶1(物质的量比),改性剂CaO加入量分别为0、1%、2%、4%、8%、16%(摩尔分数) 。称取一定量的Cu(NO3)2·3H2O、Zn(NO3)2·6H2O、Zr(NO3 )4 ·5H2O、Ca(NO3)2·4H2O(AR,国药集团化学试剂有限公司)溶于去离子水中,配制成金属离子浓度为1.0mol/L 的硝酸盐混合溶液,记为A;另称取一定量的Na2CO3(AR,国药集团化学试剂有限公司)溶于去离子水中,配制成浓度为1.1mol/L的溶液,记为B。70℃搅拌下,将A与B同时滴加,调节B滴速以控制溶液pH值为7.0;滴加完后,80℃搅拌老化1.5h,老化母液冷却至室温,抽滤、洗涤,110℃干燥24h,得到前驱体,前驱体于350℃焙烧5h,得到催化剂母体,研磨、压片、造粒,得到20-40目的催化剂母体颗粒待用。研究将110℃干燥24h后样品称为催化剂前驱体,标记为P-blank、P-CaO1%、P-CaO2%、P-CaO4%、P-CaO8%、P-CaO16%;催化剂前驱体焙烧后得到的样品为催化剂母体,标记为M-blank、M-CaO1%、M-CaO2%、M-CaO4%、M-CaO8%、M-CaO16%;催化剂母体经H2还原活化后为目标催化剂,标记为CZZblank、CZZCaO1%、CZZCaO2%、CZZCaO4%、CZZCaO8%、CZZCaO16%。
1.2 催化剂的表征
N2吸附脱附在美国康塔(Quantanchrome)公司Autosorb-iQ-C分析仪上进行。样品在300℃脱气处理5h,以高纯N2为吸附质,在液氮温度(77.3K)下进行测定,计算机采集数据。
XRD表征在日本理学D/maxrC 型X射线衍射仪上进行,Cu Kα 射线(λ=0.154056nm),扫描速率8(°)/min,石墨单色管,管电压和管电流分别为40kV和100mA,步长0.02°,10°-90°扫描。
热重-微商热重(TG-DTG)实验在德国耐驰STA 449F3型热分析仪上进行,催化剂前驱体10mg,无水空气流量50mL/min,Ar 流量20mL/min,升温速率10℃/min。
FT-IR实验在德国BRUKER VERTEX70 红外光谱仪上进行,采用RT-DLaTGS检测器,扫描次数为32,分辨率为12cm-1,测定催化剂前驱体官能团结构,所测样品由催化剂前驱体与KBr以质量比2∶50混合压片制得。
XPS在日本PHI 5000 VersaProbe-II上进行,测定催化剂元素结构,铝靶Kα发射(hv=1486.6eV),X射线阳极功率为250W,电压为14.0kV。
H2-TPR在美国康塔ChemBET Pulsar & TPR/TPD 全自动程序升温化学吸附仪上进行,热导检测器(TCD)检测。催化剂用量20mg,还原气为10%H2-90%He 混合气,气体流量20mL/min,升温速率10℃/min。
铜的比表面积和铜的分散度在美国康塔ChemBET Pulsar & TPR/TPD 全自动程序升温化学吸附仪上根据以下三个反应进行测定:
反应(1)消耗的H2记为n1,反应(3)消耗的H2记为n2,铜的分散度为:
铜的比表面积为
式中,N为阿伏伽德罗常数(6.02×1023原子数/mol),W为表征催化剂的质量,1.4×1019为每平方米面积所具有的铜原子数。具体表征步骤为:催化剂用量20mg,以He作为载气,流量为20mL/min。催化剂先在H2流中以10℃/min程序升温至600℃,消耗H2记为n1,在He中降温至60℃,切换成N2O(10%N2O-90%Ar)氧化60min,再切换为H2以10℃/min程序升温至600℃,此时消耗H2记为n2。
程序升温脱附由美国康塔ChemBET Pulsar & TPR/TPD 全自动程序升温化学吸附仪上进行。催化剂用量80mg,以He作为载气,流量为20mL/min。催化剂先在H2流中升温至280℃,还原30min,再切换成He吹扫25min,降温至60℃,切换为CO2或者NH3吸附至饱和,用He吹扫25min,开始程序升温脱附,以10℃/min升温至900℃。
SEM表征在荷兰FEINova NanoSEM 450场发射扫描电子显微镜上进行。TEM表征在日本电子JEM-2100透射电子显微镜上进行。
1.3 催化剂的活性评价
用自制高温高压微催化反应系统进行催化剂活性评价。将2.0g 20-40目催化剂装入反应管(内径9mm,长340mm),两端填充等粒度石英砂。反应前催化剂用工业氢在280℃下还原12h,冷却至室温,切换为反应原料气。原料气组成VH2∶VCO2=3∶1,在 250℃、3.0MPa、空速为3000h-1条件下反应7h。在反应前,通过气体流量计调节混合气体流量,反应后尾气流量用皂膜流量计监测。测定时,反应尾气直接进入Agilent6890气相色谱仪分析测试。气相色谱仪装有HP-MOLESIEVE(30m×0.535mm×50.00μm)毛细色谱柱和HP-PLOT/Q(30m×0.535mm×40.00μm)毛细色谱柱,CO、CO2、N2、H2采用TCD测定,CH4、CH3OH 等有机物采用FID测定,用气相色谱仪分析与产物浓度相近但不同的CO、CO2、CH4混合标气、不同浓度的甲醇溶液,得出CO、CO2、CH4、CH3OH浓度对信号值的标准曲线。根据检测出的CO、CO2、CH4、CH3OH信号值,通过标准曲线计算出CO2 转化率、甲醇选择性。
2 结果与讨论
2.1 催化剂前驱体表征
2.2 催化剂母体表征
2.3 催化剂的表征
2.1.3 FT-IR表征
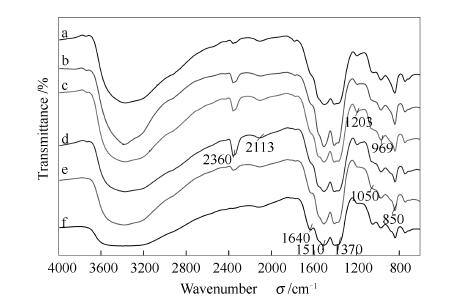
图 3为催化剂前驱体FT-IR谱图,2800-3800cm3-1为前驱体内-OH伸缩振动吸收峰,2113与2360cm-1为空气中CO2伸缩振动吸收峰,1640cm-1为催化剂前驱体少量吸附水-OH弯曲振动吸收峰,1100-1600cm-1区域内四个吸收峰(1550、1510、1403、1370cm-1)是由孔雀石或绿铜锌矿相中碳酸根离子C-O不对称伸缩振动产生。850cm-1是由孔雀石或绿铜锌矿碳酸根离子振动吸收引起。指纹区1050cm-1为孔雀石-OH振动特征吸收峰,969、1203cm-1为绿铜锌矿M-O-H弯曲振动引起。特征吸收峰[18]2800-3800cm-1区域-OH吸收峰宽泛是因为前驱体存在具有-OH的不同晶型,引起-OH化学环境不同,不同-OH振动吸收峰相互叠加。随着CaO量增加,-OH伸缩振动吸收峰变得尖锐,表明前驱体含-OH的晶相增加。1550、1510、1403、1370cm-1四个吸收峰随着CaO量增加强度逐渐增强,说明前驱体含CO2-晶相增加。Ellas等[21]发现,1050cm-1为孔雀石相特征吸收峰,969、1203cm-1为绿铜锌矿相特征吸收峰,表明前驱体中一定存在孔雀石相与绿铜锌矿相,这与前驱体XRD的表征结果吻合。添加CaO后,969、1203cm-1吸收峰增强,CaO为2%时吸收峰最强,进一步说明CaO促进绿铜锌矿相形成,且CaO为2%时绿铜锌矿相最多。由于Ca掺入了Cu2(CO3)(OH)2和 Zn3Cu2(OH)6(CO3)2中形成了Ca-OCO2,改变了C-O键强度,使得1050、969、1203cm-1三处吸收峰分别红移至1065、980、1211cm-1处,且随着掺入量增加,峰位红移量越大[22, 23]。
图 3
催化剂前驱体FT-IR 谱图
Figure 3.
FT-IR profiles of the catalyst precursors
 图 4
催化剂母体XRD谱图
Figure 4.
XRD patterns of the precatalysts
图 4
催化剂母体XRD谱图
Figure 4.
XRD patterns of the precatalysts
2.1.2 热分析
为研究催化剂性能,分析比较了不同催化剂前驱体在空气中的热分解行为。图 2为催化剂前驱体热分析谱图,各催化剂前驱体在60-200℃有少量失重,对应为前驱体物理吸附水脱出。各催化剂前驱体在200-400℃、400-560℃有明显失重峰。其中,200-400℃为催化剂前驱体中绿铜锌矿、孔雀石、水锌矿脱羟基脱羰基,400-560℃归结为高温碳酸盐分解为金属氧化物脱附峰。以P-CaO2%组为例,在332.8℃绿铜锌矿分解如下:
 图 2
催化剂前驱体热分析曲线
Figure 2.
Curves of thermal analysis of the catalyst precursors
图 2
催化剂前驱体热分析曲线
Figure 2.
Curves of thermal analysis of the catalyst precursors
512.8℃高温碳酸盐分解如下:
60-200℃脱去物理水失重4.98%,200-400℃绿铜锌矿、孔雀石、水锌矿脱羟基脱羰基失重为16.16%,这阶段失重最多。400-560℃高温碳酸盐分解失重5.6%,催化剂各阶段失重情况见表 1。
 表 1
催化剂前驱体热分析
Table 1.
Thermal analysis data of the catalyst precursors
表 1
催化剂前驱体热分析
Table 1.
Thermal analysis data of the catalyst precursors
200-400℃ 13 308.9 13.63 316.2 16.16 332.8 15.77 313.1,373.6 15.25 319.6 表 1 催化剂前驱体热分析
Table 1. Thermal analysis data of the catalyst precursorsBehrens等[18]研究表明,前驱体只有包含Cu/Zn混合共存的绿铜锌矿,热分析时才会出现高温分解阶段。前驱体经过脱水、脱CO2,生成“高温碳酸盐”。 “高温碳酸盐”在此高温分解阶段分解脱除CO2。实验体系高温碳酸盐主要为CuCO3、ZnCO3,其局限于CuO-ZnO界面之间,分解温度在400℃以上。由催化剂前驱体热分析数据表(表 1)可知,与空白组比较,加入CaO助剂后,各组在200-400℃阶段失重增加,P-CaO2%组失重最多16.16%,即更多碱式碳酸盐发生了分解;与空白组相比,各组在400-560℃阶段,对应失重温度升高,热稳定性增强,高温碳酸盐分解失重增加,其中,P-CaO2%组高温碳酸盐分解温度最高为512.8℃,分解量最多,失重5.6%,表明P-CaO2%组高温碳酸盐含量最高,前驱体含有更多绿铜锌矿。结合前驱体XRD谱图(图 1),CaO加入增强了Cu-Zn间相互作用,促进了绿铜锌矿形成。绿铜锌矿分解产生CuCO3、ZnCO3,在前驱体焙烧温度下,CuCO3、ZnCO3未完全分解。Bems等[19]发现,未完全分解高温碳酸盐促进催化反应高活性位点的生成,抑制催化剂金属氧化物晶型增长。Baltes等认为[20],催化剂母体残余的高温碳酸盐,可阻止催化剂结构的断裂破碎,形成更多铜锌相互作用的小颗粒。结合表 2,CuO、ZnO颗粒随着CuCO3、ZnCO3含量增加而变小。结合催化剂评价表 4,焙烧后催化剂中未完全分解的CuCO3、ZnCO3含量越高,其催化效果越好,表明形成更多高活性位点。
2.1.1 XRD表征
图 1为催化剂前驱体XRD谱图。由图 1可知,催化剂前驱体衍射峰均比较弥散,表明前驱体中各物相结晶并不完整。各催化剂前驱体中均出现了13.0°、24.2°、34.3°的绿铜锌矿相Zn3Cu2(OH)6(CO3)2特征衍射峰(PDF17-0743),13.1°、24.3°、32.8°、36.2°的水锌矿相Zn5(CO3)2(OH)6特征衍射峰(PDF72-1100),14.8°、17.6°、24.1°、32.3°的孔雀石相Cu2CO3(OH)2特征衍射峰(PDF41-1390)。CaO加入有利于前驱体形成孔雀石相Cu2CO3(OH)2,水锌矿相Zn5(CO3)2(OH)6,绿铜锌矿相Zn3Cu2(OH)6(CO3)2。当CaO加入量为2%时,绿铜锌矿相特征峰34.3°峰值最强,表明绿铜锌矿相形成最好。绿铜锌矿相Zn3Cu2 (OH)6(CO3)2是由Cu2+部分取代水锌矿相Zn5(CO3)2(OH)6中Zn2+形成。这种部分取代增强了Cu-Zn之间相互作用,使得CuO、ZnO颗粒更稳定[15]。研究表明,Cu-ZnO-ZrO2催化剂体系,其活性与前驱体物相结构密切相关,因为前驱体具有“化学记忆效应”,即前驱体决定氧化中间产物的氧化还原温度,以此控制Cu/ZnO基催化剂的形态和缺陷[16]。Li等[17]研究发现,绿铜锌矿相Zn3Cu2(OH)6(CO3)2焙烧后,CuO、ZnO相互聚集,还原后CuO、ZnO界面之间形成Cu-ZnO协同作用活性位,提高了催化剂活性。适量CaO改性,催化剂前驱体形成更多绿铜锌矿相Zn3Cu2(OH)6(CO3)2,焙烧后形成的CuO、ZnO颗粒变小,衍射峰变弱(参见图 4),界面协同作用活性位数量增加,催化剂活性提高。
 图 1
催化剂前驱体的XRD谱图
Figure 1.
XRD patterns of the catalyst precursors
图 1
催化剂前驱体的XRD谱图
Figure 1.
XRD patterns of the catalyst precursors
2.2.3 氮气吸附脱附表征
图 7为催化剂母体N2吸附脱附曲线。由图 7可知,根据IUPAC分类,吸附脱附曲线属于II型等温线,催化剂材料表面作用力弱,为介孔结构,具有明显的H3型迟滞环,为裂隙孔。迟滞环先增大后有所减小,表明平均孔径先增加后变小。图 8为催化剂母体孔径分布图。由图 8可知,所有催化剂孔径为10nm的孔数较多,分布比较均匀,表明共沉淀法可得到孔径均匀的介孔材料[26]。随着CaO添加量增加,孔径为10nm的孔数先增加,1%时最多,后逐渐减少。催化剂中铜颗粒粒径为7.4nm,可较易进入孔道中,提高Cu的分散性,获得高催化活性。表 2为催化剂母体孔结构参数。由表 2可知,添加CaO后催化剂的BET比表面积和孔容均有所增加,结合孔径分布图(图 8),表明CaO可促进孔的生成,使得比表面积增大,并且可调控孔的大小,使得孔径变得规整。CaO含量为4%时,比表面积最大100.6m2/g,孔容最大0.322mL/g。CaO量继续增加,比表面积和孔容均变小,孔径规整度下降,过多CaO进入催化剂,阻塞孔道,造成孔减少,变得不规整,比表面积减小。
 图 7
催化剂母体氮气吸附脱附曲线
Figure 7.
Nitrogen adsorption desorption curves of the catalysts
图 7
催化剂母体氮气吸附脱附曲线
Figure 7.
Nitrogen adsorption desorption curves of the catalysts
 图 8
催化剂母体孔径分布
Figure 8.
Pore size distribution of the catalysts
图 8
催化剂母体孔径分布
Figure 8.
Pore size distribution of the catalysts
2.2.2 SEM表征
图 6为催化剂母体SEM照片。由图 6可知,各催化剂母体结晶状态都不理想,ZnO颗粒与CuO颗粒难以区分,与催化剂母体XRD各衍射峰较为弥散相吻合。空白组催化剂母体呈絮块状,有一定团聚。加入CaO后,催化剂母体呈现明显颗粒状,且分布均匀,证明分散性提高。
 图 6
催化剂母体SEM照片
Figure 6.
SEM images of the catalysts
图 6
催化剂母体SEM照片
Figure 6.
SEM images of the catalysts
2.2.4 XPS表征
图 9为催化剂母体Cu 2p的XPS谱图。由图 9可知,所有焙烧后的催化剂只有Cu2+一种价态,存在932.2、941、952.2、961eV四个峰,峰型与CuO标准谱相比,941、961eV两个卫星峰较弱,由存在的高温碳酸铜引起[27]。由于加入了ZnO,与标准CuO谱图相比,催化剂CuO的谱图两个主峰键能减少1.4eV(标准CuO 2p3/2 933.6eV、2p1/2 953.6eV)表明Cu-Zn协同作用使得CuO键能降低,更易被H2还原。添加CaO后,CuO 2p3/2峰变得狭窄尖锐,峰位向低键能方向移动,说明CaO促进CuO向表面聚集,增强了Cu-Zn协同作用,降低了Cu-O键能,使催化剂母体更易被还原(与H2-TPR表征吻合),结合催化剂评价表 4,易还原的催化剂母体具有更高催化活性。图 10为催化剂母体XPS Zn 2p谱图。由图 10可知,只存在Zn2+价态,2p1/2峰位1044eV、2p3/2峰位1021eV比标准ZnO(2p3/2 1021.7eV、2p1/2 1044.7eV)降低0.7eV,使得ZnO更容易吸附CO2。CaO促进表面ZnO的生成,使得2p3/2峰强度变大,增强了CuO对ZnO的作用,峰位右移,使得表面ZnO更容易吸附CO2(与CO2-TPD表征吻合)。
 图 9
催化剂母体XPS Cu 2p谱图
Figure 9.
Cu 2p XPS patterns of the catalysts
图 9
催化剂母体XPS Cu 2p谱图
Figure 9.
Cu 2p XPS patterns of the catalysts
 图 10
催化剂母体XPS Zn 2p谱图
Figure 10.
Zn 2p XPS patterns of the catalysts
图 10
催化剂母体XPS Zn 2p谱图
Figure 10.
Zn 2p XPS patterns of the catalysts
2.2.1 XRD表征
图 4为催化剂母体XRD谱图。由图 4可知,31.7°、34.3°、36.1°、56.5°、68.0°为ZnO衍射峰(PDF75-1526),35.3°、38.5°为CuO衍射峰(PDF80-1916)[24]。由于ZrO2含量少,其以微晶或者无定形态较好地分散在ZnO、CuO颗粒中,XRD谱图没有出现ZrO2衍射峰。加入CaO,ZnO、CuO的衍射峰减弱,说明抑制了CuO、ZnO颗粒长大,使其分散更均匀。在36.1°ZnO衍射峰与35.3°CuO衍射峰有部分重叠,这是由于Cu2+(0.073nm)和Zn2+(0.074nm)离子半径相近,所带电荷一样,发生同晶取代形成固溶体引起,使得Cu-Zn协同作用增强[25]。根据谢乐公式计算出CuO、ZnO颗粒粒径,具体见表 2。
由表 2可知,随着CaO增至2%,CuO与ZnO颗粒粒径分别由10.9与16.8nm减少至6.0与6.3nm,CaO量继续增至16%,此时,CuO与ZnO颗粒粒径反而增大至10.7与16.2nm,进一步说明CaO可抑制颗粒增长,随着CaO量增加,抑制作用减弱。结合前驱体XRD谱图,加入CaO形成了更多绿铜锌矿相,焙烧后形成更多高温碳酸盐,提高了其热稳定性[20],使得晶粒增长得到抑制。图 5为催化剂母体280℃氢气还原后的XRD谱图。由图 5可知,加入2%CaO后,Cu(111)与Cu(200)峰值信号增强,峰位变得宽泛。由谢乐公式计算,加入2%CaO铜的晶粒粒径为4.6nm,不加助剂铜的晶粒粒径为7.4nm,进一步说明钙助剂提高了催化剂的稳定性,抑制了Cu颗粒的增长。
表 2
催化剂母体孔结构参数
Table 2.
Pore structure parameters of the catalysts
v/(mL·g-1) 0.237 0.305 0.321 0.322 0.298 0.252 d(CuO) /nm 10.9 7.3 6.0 9.0 10.5 10.7 表 2 催化剂母体孔结构参数
Table 2. Pore structure parameters of the catalysts 图 5
催化剂的XRD谱图
Figure 5.
XRD patterns of the catalysts
图 5
催化剂的XRD谱图
Figure 5.
XRD patterns of the catalysts
2.2.5 FT-IR表征
图 11为催化剂母体红外光谱谱图,在1700-1100cm-1出现了由于碳酸根阴离子中C-O非对称伸缩振动引起的1612、1458、1381cm-1三个裂分吸收峰。833cm-1吸收峰是由于面外O-C-O弯曲振动引起的[18]。C-O非对称伸缩振动吸收峰表明,催化剂前驱体350℃焙烧后,得到的催化剂母体中含有高温碳酸盐,进一步印证热分析中高温碳酸盐在400℃左右开始分解。随着CaO加入,四个峰分别红移至1643、1488、1403、850cm-1处,且吸收强度增大,表明高温碳酸盐含量增加,CaO助剂促进高温碳酸盐生成,提高其热稳定性。图 12为不同焙烧温度催化剂母体红外光谱谱图。由图 12可知,500℃焙烧时,1612、1458、1381cm-1三处吸收峰与350℃焙烧相比,强度变弱,更多高温碳酸盐发生了分解,进一步验证催化剂母体中存在高温碳酸盐。
 图 11
催化剂母体红外光谱谱图
Figure 11.
FT-IR profiles of the catalysts
图 11
催化剂母体红外光谱谱图
Figure 11.
FT-IR profiles of the catalysts
 图 12
不同焙烧温度催化剂母体红外光谱谱图
Figure 12.
FT-IR profiles of the catalysts calcined at different temperatures
图 12
不同焙烧温度催化剂母体红外光谱谱图
Figure 12.
FT-IR profiles of the catalysts calcined at different temperatures
2.3.1 H2-TPR表征
图 13为催化剂H2-TPR谱图,α还原峰(230℃左右)为催化剂母体表面CuO还原,β还原峰(267℃左右)对应体相CuO还原。加入CaO后,α、β峰的最大还原温度均降低,表明CuO更易被H2还原[28-30]。结合催化剂XRD谱图(图 5),相同还原条件下,不加Ca助剂,38.5℃CuO峰依然存在;添加Ca助剂,38.5℃CuO(111)峰消失,Cu(111)峰变强,进一步说明CaO使CuO更易还原。由表 3中α/(α+β)可看出,随着CaO增加,表面CuO还原峰面积占总CuO还原峰面积比例由0.29增加至0.35,后又下降,说明表面分散CuO的数量先增加后有所减少。结合催化剂母体XRD谱图(图 4),加入CaO,使得CuO颗粒粒径变小,提高了表面CuO分散度,过量CaO占据了CuO位置或表面缺陷位,使CuO难以还原。
 图 13
催化剂的H2-TPR谱图
Figure 13.
H2-TPR profiles of the catalysts
表 3
催化剂H2-TPR还原峰数据
Table 3.
H2-TPR reduction peaks of the catalysts
图 13
催化剂的H2-TPR谱图
Figure 13.
H2-TPR profiles of the catalysts
表 3
催化剂H2-TPR还原峰数据
Table 3.
H2-TPR reduction peaks of the catalysts
CZZCaO1% 2487 229 5704 260.6 0.30 CZZCaO2% 2871 226.9 5277 258.2 0.35 CZZCaO4% 2359 233 5565 264 0.30 CZZCaO8% 1963 217.4 5880 251.1 0.25 表 3 催化剂H2-TPR还原峰数据
Table 3. H2-TPR reduction peaks of the catalysts2.3.4 NH3-TPD表征
催化剂载体表面酸度直接影响金属与载体相互作用的程度和活性金属的分散状态[34]。NH3-TPD可表征催化剂表面酸性位点分布情况。图 16为催化剂母体NH3-TPD谱图。由图 16可知,α峰(195℃)为催化剂弱酸中心NH3脱附,β峰(442℃)、γ峰(616℃)为中强酸、强酸中心的NH3脱附。催化剂中存在ZrO2,Jung认为[35],由于ZrO2表面羟基的两性特性,其表面存在路易斯酸性位点,可吸收NH3。Samson等[36]发现,ZrO2中存在氧空位,氧空位处于缺电子状态,为催化剂酸性中心,可吸附NH3和CO2一类给电子分子。随着CaO含量增加,ZrO2分散性变好,ZrO2表面路易斯酸性增强,α峰、β峰、γ峰温度分别增至265、469、654℃,ZrO2吸附NH3量增加。
 图 16
催化剂的NH3-TPD谱图
Figure 16.
NH3-TPD profiles of the catalysts
图 16
催化剂的NH3-TPD谱图
Figure 16.
NH3-TPD profiles of the catalysts
2.3.2 TEM表征
图 14为催化剂的TEM照片。由图 14可以看出,ZnO为六方晶相,Cu为立方晶相。空白组催化剂,ZnO颗粒与Cu颗粒粒径相差明显,ZnO颗粒与Cu颗粒各自有一定团聚。加入CaO后,ZnO和Cu的颗粒均变小,Cu的颗粒覆盖在ZnO颗粒周围,ZnO和Cu接触面增加,Cu分散性变好,与催化剂XRD表征吻合(图 5),进一步证明CaO提高了催化剂稳定性,促进了Cu-Zn相互作用。
 图 14
催化剂的TEM照片
Figure 14.
TEM images of the catalysts
图 14
催化剂的TEM照片
Figure 14.
TEM images of the catalysts
2.3.3 CO2-TPD表征
图 15为催化剂母体CO2-TPD谱图。α脱附峰(132℃)为表面Cu对CO2线性吸附,线性吸附结构为O=C=O-M(M代表金属)较易脱附。β脱附峰(407℃)、γ脱附峰(604℃)分别为表面Cu、受ZnO作用的表面Cu对CO2桥式活化吸附,吸附结构(M为金属)难以脱附[31]。Hattori[32]认为,线式吸附为单吸附位,吸附强度较弱;桥式吸附为双吸附位,吸附强度较强。随着CaO加入,α脱附峰、β脱附峰、γ脱附峰分别移至138、452、663℃。结合H2-TPR结果,CaO促进表面CuO生成,结合XPS表征,CaO促进表面ZnO-CuO相互作用。CaO使得表面Cu对CO2线性吸附、表面Cu对CO2桥式吸附、受ZnO作用的表面Cu对CO2桥式吸附均增强,使得脱附峰温度升高。CaO同时增强了催化剂表面碱性,使其可在较低温度下吸附CO2,较易脱附。但CaO总体含量不高,ZnO、CuO对CO2的吸附作用应该占主导。Arena等[33]发现,ZnO和ZrO2均可吸附CO2生成甲酸盐,ZrO2表面具有碱性位点。ZrO2以无定形态分散于催化剂中,CaO可能改善ZrO2的分散性,使得碱性位点增加,CO2吸附增强。CaO增多后,表面ZnO-CuO相互作用增强,高温脱附峰β、γ峰面积增加,且γ峰出现小肩峰,说明线式吸附向桥式吸附转变,对CO2吸附增强。对CO2过强吸附力将导致催化反应阶段CO2难以脱附,与H2反应减弱,结合催化剂评价结果(表 4),印证了过多CaO加入使得CO2和H2催化合成甲醇活性下降。
 图 15
催化剂的CO2-TPD谱图
Figure 15.
CO2-TPD profiles of the catalysts
图 15
催化剂的CO2-TPD谱图
Figure 15.
CO2-TPD profiles of the catalysts
2.3.5 催化剂的活性评价
表 4为催化剂的催化性能。由表 4可知,添加CaO后,铜比表面积增大,铜的分散性增加,CO2转化率、甲醇选择性、甲醇收率都增大。结合XPS谱图,CaO增强了催化剂表面Cu-Zn协同作用,使得催化剂具有高活性[17, 19]。同时,CaO可提高催化剂的热稳定性,防止CuO烧结,使得CuO颗粒粒径变小,少量Cu2+溶解在ZnO晶格中,使得Cu分散度增加[20]。结合催化剂母体SEM照片(图 6),加入CaO后,催化剂由絮块状变为颗粒状,CuO颗粒分散变得均匀。结合催化剂TEM照片(图 14),加入CaO后,ZnO和Cu的颗粒粒径变小,分散性变好。随着CaO量增加,铜比表面积、铜分散度、CO2转化率、甲醇选择性、甲醇收率均先增加后减少,在CaO量为2%时,铜比表面积79.3m2/g、铜分散度34.8%、CO2转化率为24.55%、甲醇选择性为19.01%、甲醇收率为0.044g/(gcat·h)均为最大。CaO添加量为2%时,Cu颗粒粒径最小,易进入ZnO晶格中,形成更多Cu-Zn固溶体。过多CaO占据了催化剂孔道和CuO的活性位,使Cu比表面积减小分散性降低,CuO与H2有效接触减少,催化活性下降。
表 4
催化剂的催化性能
Table 4.
Reaction performance of the catalysts
CZZCaO1% 16.08 25.1 0.038 50.86 20.5 CZZCaO2% 19.01 24.55 0.044 79.3 34.8 CZZCaO4% 14.09 24.44 0.032 14.9 7.1 CZZCaO8% 13.69 24.07 0.026 27.8 11.6 A(Cu): the copperspecific surface area; D(Cu): the dispersion degree of copper;
reaction conditions: t= 250℃,p=3MPa,H2 /CO2= 3∶1(volume ratio) and SV=3000mL/(g·h)表 4 催化剂的催化性能
Table 4. Reaction performance of the catalysts3 结 论
用CaO作为助剂,采用并流共沉淀法制备了不同CaO添加量Cu-ZnO-ZrO2催化剂。实验表明,添加CaO后催化剂前驱体形成更多绿铜锌矿相,催化剂中高温碳酸盐增加;提高了催化剂热稳定性,CuO颗粒粒径变小,分散性增加,更易进入ZnO晶格中形成固溶体;增强了Cu-Zn协同作用,提高了催化剂活性;增强了催化剂路易斯酸性和表面碱性;增大了催化剂比表面积。催化剂活性受到表面酸碱性、铜比表面积、Cu-Zn协同作用和铜分散性共同影响。过量CaO占据催化剂孔道和覆盖表面活性位,使催化剂路易斯酸性和表面碱性过强,CuO与H2有效接触减少,CO2难以脱附,催化活性下降。
-
-
[1]
STEWART C, HESSAMI M. A study of methods of carbon dioxide capture and sequestration-the sustainability of a photosynthetic bioreactor approach[J]. Energy Convers Manage, 2005, 46(3): 403-420. doi: 10.1016/j.enconman.2004.03.009
-
[2]
LI L, ZHAO N, WEI W, SUN Y H. A review of research progress on CO2 capture, storage, and utilization in Chinese academy of sciences[J]. Fuel, 2013, 108: 112-130. doi: 10.1016/j.fuel.2011.08.022
-
[3]
ARESTA M, DIBENEDETTO A. Utilization of CO2 as a chemical feedstock: Opportunities and challenges[J]. Dalton Trans, 2007, 28: 2975-2992.
-
[4]
SHAMSUL N S, KAMARUDIN S K, RAHMAN N A, KOFLI N T. An overview on the production of bio-methanol as potential renewable energy[J]. Renew Sust Energy Rev, 2014, 33(2): 578-588.
-
[5]
BAIKER A. Utilization of carbon dioxide in heterogeneous catalytic synthesis[J]. Appl Organomet Chem, 2000, 14(12): 751-762. doi: 10.1002/(ISSN)1099-0739
-
[6]
NATESAKHAWAT S, LEKSE J W, BALTRUS J P, OHODNICKI JR P R, HOWARD B H, DENG X Y, MATRANGA C. Active sites and structure-activity relationships of copper-based catalysts for carbon dioxide hydrogenation to methanol[J]. ACS Catal, 2012, 2(8): 1667-1676. doi: 10.1021/cs300008g
-
[7]
LI C M, YUAN X D, FUJIMOTO K. Development of highly stable catalyst for methanol synthesis from carbon dioxide[J]. Appl Catal A: Gen, 2014, 469: 306-311.
-
[8]
MA Y, SUN Q, WU D, FAN W H, ZHANG Y L, DENG J F. A practical approach for the preparation of high activity Cu/ZnO/ZrO2 catalyst for methanol synthesis from CO2 hydrogenation[J]. Appl Catal A: Gen, 1998, 171(1): 45-55. doi: 10.1016/S0926-860X(98)00079-9
-
[9]
SLOCZYNSKI J, GRABOWSKI R, OLSZEWSKI P, KOZLOWSKA A, STOCH J, LACHOWSKA M, SKRZYPEK J. Effect of metal oxide additives on the activity and stability of Cu/ZnO/ZrO2 catalysts in the synthesis of methanol from CO2 and H2[J]. Appl Catal A: Gen, 2006, 310(8): 127-137.
-
[10]
SLOCZYNSKI J, GRABOWSKI R, KOZLOWSKA A, OLSZEWSKI P, LACHOWSKA M, SKRZYPEK J, STOCH J. Effect of Mg and Mn oxide additions on structural and adsorptive properties of Cu/ZnO/ZrO2 catalysts for the methanol synthesis from CO2[J]. Appl Catal A: Gen, 2003, 249(1): 129-138. doi: 10.1016/S0926-860X(03)00191-1
-
[11]
BAN H Y, LI C M, ASAMI K J, FUJIMOTO K. Influence of rare-earth elements (La, Ce, Nd and Pr) on the performance of Cu/Zn/Zr catalyst for CH3OH synthesis from CO2[J]. Catal Commun, 2014, 54: 50-54. doi: 10.1016/j.catcom.2014.05.014
-
[12]
TAN L, YANG G H, YONEYAMA Y, KOU Y L, TAN Y H, VITIDSANT T, TSUBAKI N. Iso-butanol direct synthesis from syngas over the alkali metals modified Cr/ZnO catalysts[J]. Appl Catal A: Gen, 2015, 505: 141-149. doi: 10.1016/j.apcata.2015.08.002
-
[13]
ZHONG C L, GUO X M, MAO D S, WANG S, WU G S, LU G Z. Effects of alkaline-earth oxides on the performance of a CuO-ZrO2 catalyst for methanol synthesis via CO2 hydrogenation[J]. RSC Adv, 2015, 5(65): 52958-52965. doi: 10.1039/C5RA06508A
-
[14]
ELIAS K F M, LUCREDIO A F, ASSAF E M. Effect of CaO addition on acid properties of Ni-Ca/Al2O3 catalysts applied to ethanol steam reforming[J]. Int J Hydrogen Energy, 2013, 38(11): 4407-4417. doi: 10.1016/j.ijhydene.2013.01.162
-
[15]
MILLAR G J, HOLM I H, UWINS P J R, DRENNAN J. Characterization of precursors to methanol synthesis catalysts Cu/ZnO system[J]. J Chem Soc Faraday, 1998, 94(4): 593-600. doi: 10.1039/a703954i
-
[16]
BEHRENS M, SCHLÖGL R. How to prepare a good Cu/ZnO catalyst or the role of solid state chemistry for the synthesis of nanostructured catalysts[J]. Z Anorg Allg Chem, 2013, 639(15): 2683-2695. doi: 10.1002/zaac.v639.15
-
[17]
LI J L, INUI T. Characterization of precursors of methanol synthesis catalysts, copper/zinc/aluminum oxides, precipitated at different pHs and temperatures[J]. Appl Catal A: Gen, 1996, 137(1): 105-117. doi: 10.1016/0926-860X(95)00284-7
-
[18]
BEHRENS M, GIRGSDIES F, TRUNSCHKE A, SCHLÖGL R. Minerals as model compounds for Cu/ZnO catalyst precursors: Structural and thermal properties and IR spectra of mineral and synthetic Zincian malachite,rosasite and aurichalcite and a catalyst precursor mixture[J]. Eur J Inorg Chem, 2009, 2009(10): 1347-1357.
-
[19]
BEMS B, SCHUR M, DASSENOY A, JUNKES H, HEREIN D, SCHLÖGL R. relations between synthesis and microstructural properties of copper/zinc hydroxycarbonates[J]. Chem Eur J, 2003, 9(9): 2039-2052. doi: 10.1002/chem.200204122
-
[20]
BALTES C, VUKOJEVI S, SCHUTH F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis[J]. J Catal, 2008, 258(2): 334-344. doi: 10.1016/j.jcat.2008.07.004
-
[21]
ELIAS F, ACHIM S, THILO L, HARALD H, INGO K. The influence of the precipitation/ageing temperature on a Cu/ZnO/ZrO2 catalyst for methanol synthesis from H2 and CO2[J]. ChemCatChem, 2014, 6(6): 1721-1730. doi: 10.1002/cctc.v6.6
-
[22]
夏王琼, 唐浩东, 林胜达, 岑亚青, 刘化章. 甲醇合成Cu/ZnO催化剂前驱体的物相转变[J]. 催化学报, 2009,30,(9): 879-884. XIA Wang-qiong, TANG Hao-dong, LIN Sheng-da, CEN Ya-qing, LIU Hua-zhang. Precursor phase transition of Cu/ZnO catalyst for methanol synthesis[J]. Chin J Catal, 2009, 30(9): 879-884.
-
[23]
STOILOVA D, KOLEVA V, VASSILEVA V. Infrared study of some synthetic phases of malachite (Cu2(OH)2CO3)-hydrozincite (Zn5(OH)6(CO3)2) series[J]. Spectrochi Acta A, 2002, 58(9): 2051-2059. doi: 10.1016/S1386-1425(01)00677-1
-
[24]
SONG F E, TAN Y S, XIE H J, ZHANG Q D, HAN Y Z. Direct synthesis of dimethylether from biomass-derived syngas over Cu-ZnO-Al2O3-ZrO2(x)/γ-Al2O3 bifunctional catalysts: Effect of Zr-loading[J]. Fuel Process Technol, 2014, 126: 88-94.
-
[25]
张强, 徐征, 千载虎. 在CuO-ZnO及CuO-ZnO-ZrO2催化剂上CO2/H2低压合成甲醇的研究[J]. 催化学报, 1989,10,(1): 22-28. ZHANG Qiang, XU Zheng, QIAN Zai-hu. The study of low pressure synthesis for methanol from CO2/H2 on CuO-ZnO and CuO-ZnO-ZrO2 catalyst[J]. Chin J Catal, 1989, 10(1): 22-28.
-
[26]
ZHAO H J, LIN M G, FANG K G, ZHOU J, LIU Z Y, ZENG G F, SUN Y H. A novel Cu-Mn/Ca-Zr catalyst for the synthesis of methyl formate from syngas[J]. RSC Adv, 2015, 5(83): 67630-67637. doi: 10.1039/C5RA13555A
-
[27]
DAI W L, SUN Q, DENG J F, WU D, SUN Y H. XPS studies of Cu/ZnO/Al2O3 ultra-fine catalysts derived by a novel gel oxalate co-precipitation for methanol synthesis by CO2+H2[J]. Appl Surf Sci, 2001, 177(3): 172-179. doi: 10.1016/S0169-4332(01)00229-X
-
[28]
GUO X M, MAO D S, LU G Z, WANG S, WU G S. Glycine-nitrate combustion synthesis of CuO-ZnO-ZrO2 catalysts for methanol synthesis from CO2 hydrogenation[J]. J Catal, 2010, 271(2): 178-185. doi: 10.1016/j.jcat.2010.01.009
-
[29]
BONURA G, CORDARO M, CANNILLA C, ARENA F, FRUSTERI F. The changing nature of the active site of Cu-Zn-Zr catalysts for the CO2 hydrogenation reaction to methanol[J]. Appl Catal B: Environ, 2014, 152: .
-
[30]
GUO X M, MAO D S, LU G Z, WANG S, WU G S. CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared via a route of solid-state reaction[J]. Catal Commun, 2011, 12(12): 1095-1098. doi: 10.1016/j.catcom.2011.03.033
-
[31]
李基涛, 张伟德, 陈明旦, 区泽棠. 铜基催化剂上CO2吸附的TPD和TPSR研究[J]. 天然气化工, 1998,23,(5): 14-17. LI Ji-tao, ZHANG Wei-de, CHEN Ming-dan, OU Ze-tang. Study of TPD and TPSR of CO2 adsorption on Cu based catalyst[J]. Nat gas Chem Eng, 1998, 23(5): 14-17.
-
[32]
HATTORI H. Heterogeneous basic catalysis[J]. Chem Rev, 1995, 95(3): 537-558. doi: 10.1021/cr00035a005
-
[33]
ARENA F, ITALIANO G, BARBERA K, BORDIGA S, BONURA G, SPADARO L, FRSTERI F. Solid-state interactions, adsorption sites and functionality of Cu-ZnO/ZrO2 catalysts in the CO2 hydrogenation to CH3OH[J]. Appl Catal A: Gen, 2008, 350(1): 16-23. doi: 10.1016/j.apcata.2008.07.028
-
[34]
徐友明, 沈本贤, 何金海, 罗锡辉. 用PASCA及NH3-TPD法表征Al2O3载体表面酸度[J]. 分析测试学报, 2006,25,(1): 41-44. XU You-ming, SHEN Ben-xian, HE Jin-hai, LUO Xi-hui. Study of surface acidity of γ-Al2O3 support by PASCA and NH3-TPD[J]. J Inst Anal, 2006, 25(1): 41-44.
-
[35]
JUNG K T, BEL A T. The effects of synthesis and pretreatment conditions on the bulk structure and surface properties of zirconia[J]. J Mol Catal A: Chem, 2000, 163(1/2): 27-42.
-
[36]
SAMSON K, ŠLIWA M, SOCHA R P, GORA-MAREK K, MUCHA D, RUTKOWSKA-ZBIK D, PAUL J F, RUGGIERO-MIKOLAJCZYK M, GRABOWSKI R, SŁOCZYNKI J. Influence of ZrO2 structure and copper electronic state on activity of Cu/ZrO2 catalysts in methanol synthesis from CO2[J]. ACS Catal, 2014, 4(10): 3730-3741.
-
[1]
-

图 3 催化剂前驱体FT-IR 谱图
Figure 3 FT-IR profiles of the catalyst precursors
a: P-CaO16%; b: P-CaO8%; c: P-CaO4%; d: P-CaO2%; e:P-CaO1%; f: P-blank

图 11 催化剂母体红外光谱谱图
Figure 11 FT-IR profiles of the catalysts
a: M-CaO16%; b: M-CaO8%; c: M-CaO4%; d: M-CaO2%; e: M-CaO1%; f: M-blank

图 12 不同焙烧温度催化剂母体红外光谱谱图
Figure 12 FT-IR profiles of the catalysts calcined at different temperatures
表 1 催化剂前驱体热分析
Table 1. Thermal analysis data of the catalyst precursors
200-400℃ 13 308.9 13.63 316.2 16.16 332.8 15.77 313.1,373.6 15.25 319.6  下载: 导出CSV
下载: 导出CSV
表 2 催化剂母体孔结构参数
Table 2. Pore structure parameters of the catalysts
v/(mL·g-1) 0.237 0.305 0.321 0.322 0.298 0.252 d(CuO) /nm 10.9 7.3 6.0 9.0 10.5 10.7
下载: 导出CSV
表 3 催化剂H2-TPR还原峰数据
Table 3. H2-TPR reduction peaks of the catalysts
CZZCaO1% 2487 229 5704 260.6 0.30 CZZCaO2% 2871 226.9 5277 258.2 0.35 CZZCaO4% 2359 233 5565 264 0.30 CZZCaO8% 1963 217.4 5880 251.1 0.25
下载: 导出CSV
表 4 催化剂的催化性能
Table 4. Reaction performance of the catalysts
CZZCaO1% 16.08 25.1 0.038 50.86 20.5 CZZCaO2% 19.01 24.55 0.044 79.3 34.8 CZZCaO4% 14.09 24.44 0.032 14.9 7.1 CZZCaO8% 13.69 24.07 0.026 27.8 11.6 A(Cu): the copperspecific surface area; D(Cu): the dispersion degree of copper;
reaction conditions: t= 250℃,p=3MPa,H2 /CO2= 3∶1(volume ratio) and SV=3000mL/(g·h)
下载: 导出CSV
-

















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 11
- 文章访问数: 3253
- HTML全文浏览量: 767
 下载:
下载:
