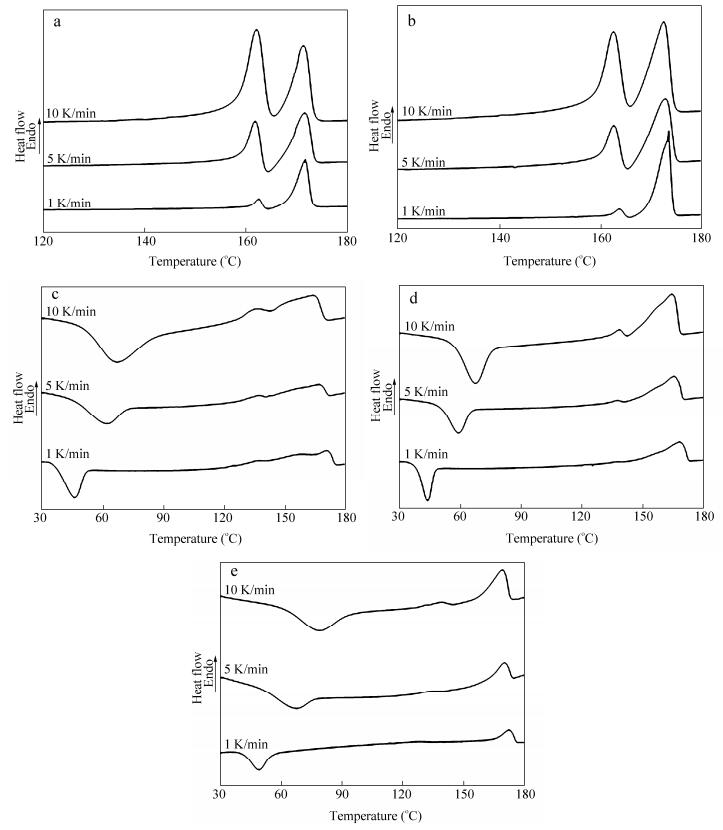
 Figure 1.
DSC melting curves of P (HB-co-HV) samples at different heating rates (1 K/min, 5 K/min, 10 K/min): (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
Figure 1.
DSC melting curves of P (HB-co-HV) samples at different heating rates (1 K/min, 5 K/min, 10 K/min): (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
Citation:
You Lv, Hao Zhu, Min-fang An, Hao-jun Xu, Li Zhang, Zong-bao Wang. Multiple Endothermic Peaks Resulted from Different Crystal Structures in an Isomorphous Copolymer Poly (3-hydroxybutyrate-co-3-hydroxyvalerate)[J]. Chinese Journal of Polymer Science,
2016, 34(12): 1510-1522.
doi:
10.1007/s10118-016-1866-5

Multiple Endothermic Peaks Resulted from Different Crystal Structures in an Isomorphous Copolymer Poly (3-hydroxybutyrate-co-3-hydroxyvalerate)
English
Multiple Endothermic Peaks Resulted from Different Crystal Structures in an Isomorphous Copolymer Poly (3-hydroxybutyrate-co-3-hydroxyvalerate)
Abstract:
The multiple endothermic peaks of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) (P (HB-co-HV)) in differential scanning calorimetry (DSC) results, as one representative phenomenon of polymer with unique cocrystallization behavior, were generally considered as the results of melting/recrystallization. In this study, wide angle X-ray diffraction (WAXD) and small angle X-ray scattering (SAXS) experiments were conducted to analyze the phenomena of multiple endothermic peaks in DSC results. The results of these analyses indicated that the multiple endotherms were mainly caused by different lamellae structures. For P (HB-co-HV) with lower HV content, it was comprised of two structures of HV total exclusion and HV partial inclusion in the crystal lamellae. For P (HB-co-HV) with higher HV content, it was also comprised of two structures of HV total inclusion and HV partial inclusion in the crystal lamellae. However, only structure with HV partial inclusion in the crystal lamellae remained existing after first melting peak for all samples.
-
Key words:
- P (HB-co-HV)
- / Isodimorphism
- / Multiple endotherms
- / Uniform lamellae
- / Sandwich lamellae
-
INTRODUCTION
Because of the characteristic of long chain, polymer crystallization requires a large number of repetitive units pack into the crystal lattice synergistically. Some copolymer with counits different from the repetitive units can be divided into two models, i.e. the uniform exclusion model and the inclusion model, according to the possibility of arranging into the crystal lattice, determined by the volume and the polarity of counits[1, 2]. However, no matter whether it can pack into the crystal lattice or not, the counits will influence the crystallization behavior of copolymer, especially in the inner structure of crystal and the amorphous layer on the crystal surface, which will lead to a remarkable crystallization kinetics effect. Flory proposed the uniform exclusion model that the crystalline phase was only composed of crystallizable units while uncrystallizable units were totally excluded[1]. Alternatively, the inclusion model allows for uncrystallizable units in amorphous and the crystalline regions at the same time, which results in the defects in crystalline, and uneven partitioning of uncrystallizable units between crystalline and amorphous phases[2]. Sanchez and Eby extended the uniform inclusion model to allow for full range partition of uncrystallizable units between the two regions[3, 4].
In most copolymers, compared to the free energy of fusion of an crystallizable unit at any crystallization condition, the chance for excess free energy upon the inclusion of an uncrystallizable unit to pack into the crystalline lattice is large. So the content of uncrystallizable units in the lamellae is generally thought to be nearly zero. However, there are a few copolymer systems where the content of uncrystallizable in the lamellae is quite large (yet it is smaller than the crystallizable content of the whole copolymer)[5]. This crystalline state is called cocrystallization, namely, isodimorphism[6]. The necessary conditions for isodimorphism of two different polymers are thought to be the similarity in the molecular structures, the crystalline lattice structures, and the crystallization rates. The first and second conditions are essential for thermodynamic stability of cocrystals, while the third one is for kinetic accessibility to form cocrystals[7].
Poly (3-hydroxybutyrate-co-3-hydroxyvalerate) (P (HB-co-HV)) is one representative thermoplastic copolymer with unique cocrystallization behavior, and it has many potential applications such as a monofilament surgical stuture and controlled drug release etc., owing to its biodegradability and biocompatibility. Further all these uses are related to its cocrystallization behavior. Poly (3-hydroxybutyrate) (PHB) and poly (3-hydroxyvalerate) (PHV) have similar crystalline structure of 21helices with fiber repeats of 0.596 and 0.556 nm, respectively[8]. The melting temperature with respect to composition of P (HB-co-HV) decreases sharply to reach a minimum value at approximately 40 mol%, and the crystalline phase develops from the PHB lattice to the PHV lattice[9-11], yet the crystallinity remains at a high level over the full range of compositions due to isodimorphism[9-12]. Consequently P (HB-co-HV) has generated much interest among the scientists of polymer physics as a special phenomenon of polymer crystallization, and related researches have lasted at least 40 years.
In previous studies, several evidences of isodimorphism in P (HB-co-HV) have been found by high-resolution solid-state 13C-NMR spectroscopy, wide angle X-ray diffraction (WAXD), differential scanning calorimetry (DSC), and polarized optical microscopy (POM)[9-11, 17]. Furthermore, the multiple DSC endotherms and a broad melting range were found in the case of P (HB-co-HV) samples. Steven et al. revealed that multiple endotherms could be explained by a distribution of crystalline sizes and the relatively broad melting endothems could be a result of different degrees of crystal imperfection[11]. Naoko et al. opined that the phenomenon was caused by polymer blends and the number of melting peaks was determined by the kinds of polymers in the mixture[18]. Naoko et al. considered that peaks arising from phase-separated structures and peaks from crystals rearranged during heating in the DSC should be distinguished, and revealed that the lower temperature peak was the melting peak of crystals formed at Tc while the higher one is the melting peak of crystals recrystallized during the heating process in the DSC measurement[19]. Mitomo and Organ and Barham predicted that the occurrence of certain high temperature endotherms for HV copolymer samples was associated with the possibility of a PHB-rich phase or the exclusion of HV units from the crystal lamellae[20-22]. Rule and Liggat observed the presence of multiple endothermic peaks in the case of copolymer but not in homopolymer, and the multiple endothermic peaks of P (HB-co-HV) in DSC results had been related to the effects of the HV units in limiting the thickness of the lamellae[23]. However, in previous studies, the measurements of crystal structure corresponding to the DSC thermal process was not executed and the particular situation of formation of multiple endothermic peaks is still an unclear issue.
In the present study, P (HB-co-HV) was chosen as a model polymer to study the isodimorphism and multiple endothermic peaks because of its inherent isodimorphic property. We have reexamined the structure of isomorphous crystals of P (HB-co-HV) with 4.9 mol%-36.2 mol% HV. In this composition range, HV units cocrystallize with HB units in the PHB lattice. The differential scanning calorimetry (DSC), wide angle X-ray diffraction (WAXD) and small angle X-ray scattering (SAXS) were combined to analyze a series of P (HB-co-HV) samples prepared in the same treatment. The results of these analyses indicated that the multiple endotherms were mainly caused by different lamellae structures. This reason of multiple endotherms had not been reported particularly in previous reports.
EXPERIMENTAL
DSC Characterization
Differential scanning calorimetry (DSC) measurements were performed with a Perkin Elmer 8000 under nitrogen atmosphere. The samples of neat P (HB-co-HV) were first heated to 190 ℃ to remove the thermal history. Subsequently, the samples were cooled to 25 ℃ at a cooling rate of 10 K/min. Finally, the samples were reheated to 190 ℃ at a heating rate of 1 K/min, 5 K/min, 10 K/min, respectively (second heating).
Analytical Methods
Small angle X-ray scattering is a well-established technique for probing the morphology of polymer and providing information describing structural features. In samples possessing a regular, ordered morphology, a correlation peak is observed in the SAXS intensity at a characteristic value of q (denoted by qmax), which is related to the separation of domains or heterogeneities in the material. In the simplest analysis, the scattering may be treated according to Bragg’s Law and hence:
where d is the average interdomain spacing and q is the scattering vector, defined as (4πsinθ)/λ where θ is the scattering angle and λ is the X-ray wavelength.
In the case of a semicrystalline polymer possessing a lamellar morphology (such as PHB and P (HB-co-HV)), which is isotropically distributed throughout the material, the Lorentz correction is applied to the SAXS intensity prior to the determination of the peak position. The Lorentz correction involves the multiplication of the intensity function by a factor of q2 and has the effect of shifting the position of a broad correlation peak to higher values of q.
A general approach for the characterization of lamellar morphologies involves the calculation of the one-dimensional correlation function, K(z), which is essentially a one-dimensional Fourier transform of the SAXS intensity[26-28]. K(z) may be defined as:
In lamellar systems, fluctuations in electron density only occur in a direction perpendicular to the plane of the lamellae. The correlation function, which is a mathematical restatement of the information contained in the SAXS pattern, provides a description of the variation in electron density as a function of distance within the sample. The interpretation of the correlation function is rather straightforward and allows direct evaluation of several structural features which describe the lamellar morphology. These include the d-spacing (or long period), the average thickness of the crystalline and amorphous layers, the (linear) crystallinity, the density difference between the two phases and the thickness of the interfacial region between the phases.
Materials
P (HB-co-HV) samples used in experiments were kindly provided by Ningbo Tian’an Biologic Material Co., Ltd. All samples were purified by solvent extraction before further investigation[24, 25]. The samples were first dissolved in chloroform and then precipitated with methanol. The precipitates were collected by filtration and dried in vacuum at room temperature for 24 h. HV contents were measured to be 4.9 mol%, 9.4 mol%, 19.4 mol%, 28.7 mol%, and 36.2 mol% by Bruker Advance III NMR spectrometer operating at 400 MHz.
WAXD and SAXS Measurements
Wide-angle X-ray diffraction (WAXD) and small-angle X-ray scattering (SAXS) measurements were carried out using Xuess System produced by Xenocs Company, France. The energy of the X-ray radiation was 50 kV, resulting in a wavelength of 0.154 nm. The size of the primary X-ray beam at the sample position was 0.3 mm × 0.3 mm. A Linkam 650 hot-stage, modified for X-ray scattering experiments, was used for all elevated temperature measurements. Two-dimensional (2D) WAXD and SAXS patterns were collected by using a Mar CCD X-ray detector (MAR345) having a resolution of 2048 × 2048 pixels. The sample-to-detector distance measured was 150 mm for WAXD and 2500 mm for SAXS. Both the WAXD and SAXS image acquisition time of each data frame was 1 h. All X-ray images were corrected for background scattering, air scattering, and beam fluctuations. The WAXD and SAXS measurement data analyses were carried out by the Fit2d software package.
The optimum sample thickness for X-ray scattering in the transmission configuration is generally considered to be 1/μ (where μ is the linear X-ray absorption coefficient). For PHB, μ was calculated to be 8.4 cm-1 at a wavelength of 0.154 nm, giving a desired thickness of around 1.2 mm. Slightly thinner samples (0.5-0.8 mm) were used in the hot-stage in order to minimize temperature fluctuations across the sample. Before experiment, all samples were inserted between aluminum plates with an aluminum spacer (0.1 mm thickness), and heated to 190 ℃ for 3 min by Linkam 650 hot-stage to remove the thermal history, then, cooled to 25 ℃ at 10 K/min, in order to correspond to the DSC procedure.
RESULTS AND DISCUSSION
There were many evidences about the multiple endotherms of P (HB-co-HV) in previous papers. It has also been found that the crystallization conditions are as important as the HV content of sample. In order to verify the phenomenon of multiple endotherms and further study the crystallization kinetics, plenty of DSC measurements were performed. Melting curves of P (HB-co-HV) samples at different heating rates were determined by DSC, as shown in Fig. 1. More details of DSC measurements such as melting point and enthalpy of fusion are listed in Table 1. Similar to the previous study, multiple endotherms can be seen in melting curves. Meanwhile, a cold-crystallization appears in P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV). Considering that HV unit has a large volume compared to HB unit and the excess free energy upon the inclusion of HV unit into the PHB crystalline lattice is larger than HB unit, it is reasonable to expect the samples with higher HV content show cold-crystallization peaks in the heating process. However, some details of thermal analysis should be noted. Different with the results observed by Inoue et al.[29], the melting curve peak changes its position when the heating rates are changed, especially for P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV). Similarly the changes of enthalpy of fusion of P (HB-co-HV) samples at different heating rates are obvious. With the scan rate decreasing, the high-temperature peak becomes larger at the expense of the low-temperature peak, indicative of a melting/recrystallization process. This is commonly observed in the thermal analysis of semicrystalline polymers. In addition, the position of each peak should be paid more attention. For P (HB-co-4.9%HV) and P (HB-co-9.4%HV), the lower melting peak is shown at around 160 ℃, which is much higher than that appears at about 140 ℃ in P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV). However, the positions of higher endotherms of all samples are similar. This result illustrates that the structures corresponding to lower endothermic peaks have significant difference, whereas they are similar in the higher ones. It can also be seen from the difference among the enthalpy of fusion. The melting enthalpy of each peak in P (HB-co-4.9%HV) and P (HB-co-9.4%HV) are much larger compared with that in higher HV content samples. However, the multiple endotherms are much intense when fast heating rate is performed. What we observed above indicates that the melting/recrystallization is not a crucial factor for multiple endotherms, while, the probable reason that leads to the multiple endotherms may result from the different HV contents in P (HB-co-HV) samples. The details should be further studied.
Figure 1.
DSC melting curves of P (HB-co-HV) samples at different heating rates (1 K/min, 5 K/min, 10 K/min): (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
 Table 1.
The melting point and enthalpy of fusion of P (HB-co-HV) samples measured at different heating rates in DSC experiment
Table 1.
The melting point and enthalpy of fusion of P (HB-co-HV) samples measured at different heating rates in DSC experiment
HV content (mol%) Heating rate (K/min) Tm1 (℃) Tm2 (℃) ΔH1 (J/g) ΔH2 (J/g) 4.9 1 162.6 171.8 14.0 58.3 5 161.9 171.6 33.3 42.5 10 162.1 171.4 41.2 28.4 9.4 1 163.5 173.3 13.9 72.8 5 162.4 172.6 26.8 55.9 10 162.3 172.3 30.2 34.7 19.4 1 136.1 170.0 1.5 27.5 5 136.3 166.6 2.4 23.5 10 134.4 163.8 3.3 19.3 28.7 1 136.1 168.0 0.3 46.3 5 137.4 165.2 1.3 41.5 10 138.0 164.1 3.4 39.6 36.2 1 127.8 173.2 0.8 16.4 5 131.7 170.0 1.0 16.3 10 138.3 169.0 1.8 16.9 Table 1. The melting point and enthalpy of fusion of P (HB-co-HV) samples measured at different heating rates in DSC experimentWAXD and SAXS are frequently used techniques for probing the structure of polymer. As it seems that multiple endotherms are caused by different structures in P (HB-co-HV) based on DSC results, WAXD and SAXS can provide more evidence about the coexistence of structure. The temperatures at which WAXD and SAXS measurements were selected on the basis of the DSC results at the heat rate of 10 K/min (Table 2). The first testing temperatures were chosen at the temperatures completing cold-crystallization for samples with high HV content in order to obtain the samples with complete crystallization; the second testing temperatures were chosen between the first and second melting temperature in order to destroy the structure corresponding to the first melting temperature. WAXD can provide the direct evidence of the structure of P (HB-co-HV) since slight cell expansion can be detected by WAXD with the inclusion of HV units in PHB lattice. WAXD curves of these copolymers are shown in Fig. 2. The diffraction peaks corresponding to the PHB lattice are observed over all range of compositions of samples[11]. The peaks shown in diffraction curves at around 13.7°, 17.2°, 20.3°, 21.8°, 22.7°, 25.9° and 27.5° correspond to (020), (110), (021), (101), (111), (121) and (040) diffraction plane of PHB, respectively. It should be noted that only PHB diffraction plane is observed in all samples, which indicates that P (HB-co-HV) used in this experiment does not have a mixed structure from different polymers as reported earlier[18]. In addition, the positions of main diffraction peaks change slightly compared with pure PHB. It can be clearly seen that the diffraction peak positions have changed after heating. The parameters of crystalline lattice calculated from WAXD results are shown in Table 3. It can be seen from Table 3 that the parameters of crystalline cell of P (HB-co-4.9%HV) and P (HB-co-9.4%HV) increase at the second testing temperature while those of P (HB-co-HV) with higher HV content decrease instead. It is easy to draw a conclusion that these slight changes result from the expansion or contraction of crystalline cells since HV units include into or exclude from the PHB lattice.
Table 2.
Testing temperature of WAXD and SAXS experiments
HV content (mol%) Testing temperature (℃) 4.9 25 160 9.4 25 161 19.4 105 145 28.7 90 140 36.2 90 145 Table 2. Testing temperature of WAXD and SAXS experiments Figure 2.
WAXD curves of P (HB-co-HV) samples after crystallization and the first structure corresponding to the lower melting point destruction: (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
Table 3.
The lattice parameters calculated from WAXD curves of P (HB-co-HV) samples
Figure 2.
WAXD curves of P (HB-co-HV) samples after crystallization and the first structure corresponding to the lower melting point destruction: (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
Table 3.
The lattice parameters calculated from WAXD curves of P (HB-co-HV) samples
HV content (mol%) Testing temperature (℃) a (nm) b (nm) 4.9 25 0.569 1.301 160 0.579 1.308 9.4 25 0.567 1.302 161 0.578 1.310 19.4 105 0.582 1.315 145 0.581 1.311 28.7 90 0.581 1.316 140 0.581 1.312 36.2 90 0.588 1.321 145 0.582 1.312 Table 3. The lattice parameters calculated from WAXD curves of P (HB-co-HV) samplesConsidering the fact that WAXD patterns reflect the statistical averages of the lattice, this result means that some structures with smaller cell disappear after heating when HV content is below 9.4 mol%, instead, some structures with larger cell are destroyed when HV content is above 19.4 mol%. In addition, the parameters of structure after heating in P (HB-co-4.9%HV) and P (HB-co-9.4%HV) are similar, most importantly the parameters are also similar to those of P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV) after heating. It reveals that all samples have the same structure after heating. These results indicate that there are two kinds of crystal structures before heating for both P (HB-co-HV) with lower and higher HV content and one kind of structure with smaller cell disappears for lower HV content P (HB-co-HV) and another kind of structure with larger cell disappears for higher HV content P (HB-co-HV). Therefore there are three kinds of structures in all the P (HB-co-HV) samples. Naoko et al. introduced two P (HB-co-HV) structural models, and they reported that P (HB-co-HV) samples with HV content below 10 mol% could be described by the sandwich lamella model in which the core was composed entirely of the HB and the HV existed at the edge of the crystal lamella (not on the amorphous layer), and P (HB-co-HV) samples with HV content above 10 mol% could be described by the uniform lamella model in which HV were uniformly distributed in the crystal lamella[5]. The similar structural transition can also be seen from our experimental results of P (HB-co-HV) with 4.9%, 9.4% and 19.4%, 28.7%, 36.2% HV content. Considering the sandwich lamella model proposed by Naoko. Y, it can be understood that only HB participates in the lamella and the HV lies on the amorphous layer when the HB chain is long for random copolymer. By summarizing the WAXD results and analyses, it is reasonable P (HB-co-HV) with lower HV content is comprised of two structures of HV total exclusion and HV partial inclusion in the crystal lamella, and that P (HB-co-HV) with higher HV content is comprised of two structures of HV total inclusion and HV partial inclusion in the crystal lamella, and only the structure with HV partial inclusion in the crystal lamella remains existing after first melting temperature.
SAXS can provide the evidence of the structure of P (HB-co-HV) since multiple crystalline layer thicknesses with the HV units inclusion or exclusion in PHB lattice. A conspicuous change in the SAXS curves is provided in Fig. 3, which illustrates the evolution of the scattering vector with increasing temperature during the heating of all of the P (HB-co-HV) copolymer samples. The SAXS scattering peak is found to grow in intensity after heating, and an obvious displacement to lower q (equivalent to higher characteristic long period spacing) of scattering peak can be observed. Meanwhile, the width of scattering peak decreases after heating. In order to represent the data of the P (HB-co-HV) samples with the correct q weighing, the scattering intensity was multiplied by q2 for data processing (Lorentz correction). The long period dac with temperature, calculated from Lorentz-corrected plots, is shown in Fig. 4. In each case, a sharp change of dac can be seen. Besides, the SAXS curves for the completely crystallized samples collected at different temperature are not symmetrical, and some of them even exhibit two scattering intensity maxima, reflecting the presence of two populations of lamellar stacks with different long spacing values (Fig. 4).
 Figure 3.
One-dimensional scattering intensity distribution profiles of SAXS experiments of P (HB-co-HV) samples without Lorentz correction: (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
Figure 3.
One-dimensional scattering intensity distribution profiles of SAXS experiments of P (HB-co-HV) samples without Lorentz correction: (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
 Figure 4.
The Lorentz-corrected (Iq2~q) SAXS curves of P (HB-co-HV) samples: (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
Figure 4.
The Lorentz-corrected (Iq2~q) SAXS curves of P (HB-co-HV) samples: (a) 4.9 mol% HV, (b) 9.4 mol% HV, (c) 19.4 mol% HV, (d) 28.7 mol% HV and (e) 36.2 mol% HV
In order to calculate the thickness of crystalline layer and further study the structure of crystal, the method of one-dimensional scattering intensity distributions was used. The one-dimensional scattering intensity distributions of crystallized P (HB-co-HV) measured before heating and the resultant correlation functions for the two populations of lamellar stacks are presented in Fig. 5. The scattering intensity distribution profile was decomposed into contributions from two populations of lamellar stacks based on Gaussian functions, as depicted in Fig. 5(a). The inset in Fig. 5(b) demonstrates how the average thickness of the long spacing (dac) and crystalline layers (dc) or the amorphous layers (da) were obtained, the long period and crystal layer thickness of different structures are shown in Table 4. It must be mentioned that it is impossible to decide whether it is the amorphous or the crystalline thickness that is read out from the correlation function without prior knowledge of crystallinity. However, according to it, the long period is accumulated by the thickness of crystalline lamella and amorphous region, the thickness of remaining crystalline lamella is larger than original crystal. It can be seen from Table 4 that there are two lamellar thickness values after P (HB-co-HV) complete crystallization for all samples, and the lamellar thickness after melting is similar to the higher thickness of that obtained after complete crystallization. The results correspond to the two kinds of structures from WAXD results, and they confirm the structural analysis described in the WAXD section based on the discussion in the following lines. The lamellar thickness calculated from SAXS data suggests that there are two kinds of lamellar after complete crystallization. The P (HB-co-HV) with lower HV content has much thicker crystal because of the long HB sequences which lead to a larger enthalpy of fusion compared with higher HV content samples. For P (HB-co-4.9%HV) and P (HB-co-9.4%HV), because of total exclusion of HV units, the lamellar thickness decreases with the decreasing of HB sequences. Meanwhile, the sandwich lamella which core is composed entirely of HB also becomes thin though the edge is thickening with the HV units increasing. However, it is still thicker than the uniform lamellar which is totally composed of HB sequences. For P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV), thinner lamellar totally include HV units was detected and it slightly changed with HV content. Meanwhile, a similar sandwich lamella could be found which is thicker with the increasing of HV content. It should be noted that the lamellar thickness with HV units total inclusion is much smaller than total exclusion and partial inclusion, the amount of total inclusion lamellar is few and lead to a thinner lamellar thickness because of not abundant content and stereo-hindrance effect. In addition, only one lamellar thickness can be observed after heating. The results illustrate that the thinner lamellae corresponding to total exclusion or inclusion lamellar disappear firstly which lead to lower melting point. Meanwhile, thicker lamellae with partial exclusion of HV units can be seen after heating. It should not be ignored that the lamellae have slight thickeness during heating because of melting/recrystallization.
Table 4.
The long spacing (dac) and lamellar thickness (dc) obtained from the one-dimensional correlation function
HV content (mol%) Testing temperature (℃) dac (nm) dc (nm) 4.9 25 9.62 4.21 13.18 5.61 160 11.99 5.76 9.4 25 8.50 3.81 9.17 5.02 161 10.89 5.19 19.4 105 6.79 2.79 8.75 3.61 145 13.14 5.54 28.7 90 7.37 3.20 8.86 3.75 140 10.40 4.25 36.2 90 7.96 3.11 10.04 4.08 145 14.60 6.14 Table 4. The long spacing (dac) and lamellar thickness (dc) obtained from the one-dimensional correlation function Figure 5.
(a) One-dimensional scattering intensity distribution of P (HB-co-HV) samples and the decomposition into two lamellar stack contributions, (b) one-dimensional correlation function derived from the two kinds of P (HB-co-HV) structures
Figure 5.
(a) One-dimensional scattering intensity distribution of P (HB-co-HV) samples and the decomposition into two lamellar stack contributions, (b) one-dimensional correlation function derived from the two kinds of P (HB-co-HV) structures
Combining the WAXD and SAXS results, it can be concluded that the lamella with lower thickness is the structure of HV totally exclude and HV totally include for P (HB-co-HV) with lower HV content (P (HB-co-4.9%HV) and P (HB-co-9.4%HV)) and higher content (P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV)), respectively, and the lamella with higher thickness is the structure with HV partial inclusion in crystal lamella. Figure 6 is a schematic representation of the models. The structure with HV partial inclusion in the crystal lamella is much thicker than that HV totally exclude, it is the reason that the structure with HV totally exclude disappears and the structure with HV partly inclusion remains after heating. For P (HB-co-4.9%HV) and P (HB-co-9.4%HV), the thicknesses of two kinds of structures decrease with increasing HV content because the average length of the HB sequence is more longer for P (HB-co-HV) with lower HV content. For P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV), the thickness decreases with increasing HV content for the structure with partial HV inclusion in crystal lamella whereas the thickness increases with increasing HV content for the structure with HV totally inclusion in crystal lamella.
 Figure 6.
Schematic drawing of three models of lamellar crystals: (a) lamellar structure with HV units total exclusion, (b) lamellar structure with HV units total inclusion, (c) sandwich lamella corresponding to HV unit partial inclusion in which the core is composed entirely of the HB unit and the counits exist only in the edge of the lamella
Figure 6.
Schematic drawing of three models of lamellar crystals: (a) lamellar structure with HV units total exclusion, (b) lamellar structure with HV units total inclusion, (c) sandwich lamella corresponding to HV unit partial inclusion in which the core is composed entirely of the HB unit and the counits exist only in the edge of the lamella
It is known that the free energy of formation of cocrystals is given by the sum of the free energy of the crystallization of the homopolymer, the excess free energy and the configurational entropy upon the inclusion the other counits into the homopolymer lattice, as well as the surface free energy. The absolute value of excess free energy of cocrystallization must be small though it is usually positive in the case of isomorphous copolymers like P (HB-co-HV). Therefore, in P (HB-co-HV) with low HV content where relatively long HB sequences are abundant and the entropy gain upon the cocrystallization is little, there must be a marked tendency for the HV counits to be excluded from the lamella. Therefore the structure of PHB lattice with total exclusion of HV units is one main structure for P (HB-co-4.9%HV) and P (HB-co-9.4%HV). On the other hand, for higher HV content samples, it is difficult to exclude HV from PHB lattice because of the abundance of HV units, whereas more HV units are packed into the lattice and the volume of cell is expanded. It is the lamella which is constituted with total HV inclusion. Therefore the structure of PHB lattice with total inclusion of HV units is one main structure for P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV). For the P (HB-co-HV) chains with medium HV, which may exist in P (HB-co-HV) samples with higher content HV and lower content HV because of random copolymer samples, the HV units may exist in the lamellar surface where the distortion of the crystalline lattice upon the inclusion of the HV units can be easily relaxed, which is coincidence with the sandwich lamellar model proposed by Naoko[5]. Therefore the structure with HV units partial inclusion in crystal lamella is the other main structure for samples with both higher HV content and lower HV content in our experiment. The analysis results are consistent with previous results that the amount of HV units in the crystals is approximately equal to two-third of the total content in the copolymer[30]. Combined with WAXD and SAXS results, it is concluded that P (HB-co-HV) with lower HV content is comprised of two structures of HV total exclusion and HV partial inclusion in the crystal lamella, and P (HB-co-HV) with higher HV content is comprised of two structures of HV total inclusion and HV partial inclusion in the crystal lamella, and only structure with HV partial inclusion in the crystal lamella still exists after first melting temperature for all samples.
By summarizing the results of DSC, WAXD and SAXS, it can be concluded that the multiple endotherms of P (HB-co-HV) with different HV content are due to the different structures formed by HV including into or excluding from the PHB crystalline cell. For P (HB-co-4.9%HV) and P (HB-co-9.4%HV), the parameters of a-axis and b-axis calculated from WAXD results suggest that the lower melting temperature corresponds to the lamellae with HV units total exclusion, and the higher melting temperature corresponds to the lamellae with partial inclusion of HV units to form sandwich model. Different from lower HV units content samples, in P (HB-co-19.4%HV), P (HB-co-28.7%HV) and P (HB-co-36.2%HV), the first endotherm peak results from the structure which totally include HV units, and the higher endotherm peak is due to the lamellae with partial inclusion of HV units to form sandwich model. Looking back at the DSC results, the tendency to change multiple endotherms of P (HB-co-HV) can be well explained by the above structure model. For P (HB-co-HV) with lower HV content, the melting temperatures increase slightly with the decreasing heating rates (Fig. 1 and Table 1) since both the crystals with HV total exclusion and sandwich lamellae can become more perfect at lower heating rate, the first enthalpy of fusion decreases with the decreasing heating rate and the second enthalpy increases (Fig. 1 and Table 1) because the crystals with HV total exclusion change into more thicker sandwich lamellae during the heating. It should be noted that difference in structure is the main reason for multiple endotherms although melting/recrystallization exists at the slow heating rate. For P (HB-co-HV) with higher HV content, the higher melting temperature and enthalpy of fusion increases with the decreasing heating rate (Fig. 1 and Table 1) because slow heating rate results in the formation of much thicker sandwich lamellae and some crystals with HV total exclusion change into sandwich lamellae, but it is confused that the lower melting temperature corresponding to the crystals decreasing HV total inclusion with the decreasing heating rate. Indeed, the higher melting temperature has shoulder peaks in the DSC curves of P (HB-co-HV) with higher HV content. Therefore, it should be pointed out that the crystal structures we discussed above is an idealized model while the true situation though similar is more complex.
CONCLUSIONS
It is clear that the multiple endotherms of P (HB-co-HV) are related to the different crystal structures. For P (HB-co-HV) samples with 4.9 mol% and 9.4 mol% HV contents, the appearance of the first endothermic peak is due to thinner crystals which are caused by the total exclusion of HV units. Meanwhile, thicker crystals, much like sandwich crystals because of partial inclusion of HV units, lead to higher melting peak. On the other hand, the reasons of multiple endotherms for P (HB-co-HV) with higher HV content are quite different. The first melting peak results from thinner crystals which include many HV units, meanwhile, the sandwich crystals left after heating are responsible for the higher endothermic peak. This work is expected to be helpful for understanding not only the multiple endothermic behavior of P (HB-co-HV) but also the structure of cocrystallization polymer. To best understand the complicated crystallization behavior of P (HB-co-HV), the investigation of in situ structural evolution of P (HB-co-HV) on heating with the synchrotron radiation source is ongoing.
-
-
[1]
Flory, P.J., Trans. Faraday Soc., 1955, 51:848 doi: 10.1039/tf9555100848
-
[2]
William, J.O., Robert, H.M. and Terry, L.B., Macromolecules, 1991, 24:6435 doi: 10.1021/ma00024a010
-
[3]
Sanchez, I.C. and Eby, R.K., Macromolecules, 1973, 77:353 http://onlinelibrary.wiley.com/resolve/reference/ADS?id=1975MaMol...8..638S
-
[4]
Sanchez, I.C. and Eby, R.K., Macromolecules, 1975, 8:638 doi: 10.1021/ma60047a012
-
[5]
Naoko, Y., Miwa, S. and Yoshio, Y., Macromolecules, 2001, 34:8953 doi: 10.1021/ma0113071
-
[6]
Allerga, G. and Bassi, I.W., Adv. Polym. Sci., 1969, 6:549 doi: 10.1007/BFb0051143
-
[7]
Naoko, Y., Akeshi, A. and Yoshio, I., Macromolecules, 2004, 37:3770 doi: 10.1021/ma049858p
-
[8]
Naoko, K., Yoshio, I., Yasuhiko, Y., Riichirô, C. and Yoshiharu, D., Macromolecules, 1990, 23:1313 doi: 10.1021/ma00207a015
-
[9]
Schlegel, H.G., Gottschalk, G. and Bastha, R.V., Nature, 1961, 191:463 doi: 10.1038/191463a0
-
[10]
Lundgren, D.G., Alper, R., Schanaitman, C. and Marchessault, R.H., J. Bacteriol., 1965, 89:245 http://onlinelibrary.wiley.com/resolve/reference/ADS?id=1975MaMol...8..638S
-
[11]
Holmes, P.A., Wright, L.F. and Collins, S.H., 1982, Eur. Pat. Appl, 0052459
-
[12]
Holmes, P.A., Wright, L.F. and Collins, S.H., 1983, Eur. Pat. Appl, 0069497
-
[13]
Cornibert, J. and Marchessault, R.H., J. Mol. Biol., 1972, 71:735 doi: 10.1016/S0022-2836(72)80035-4
-
[14]
Yokouchi, M., Chatani, Y., Tadokoro, H., Teranishi, K. and Tani, H., Polymer, 1973, 14:267 doi: 10.1016/0032-3861(73)90087-6
-
[15]
Yokouchi, M., Chatani, Y., Tadokoro, H. and Tani, H., Polym. J., 1974, 6:248 doi: 10.1295/polymj.6.248
-
[16]
Mitomo, H., Morishita, N. and Doi, Y., Macromolecules, 1993, 26:5809 doi: 10.1021/ma00073a041
-
[17]
Yamada, S., Wang, L., Asakawa, N., Yoshie, N. and Inoue, Y., Macromolecules, 2001, 34:4659 doi: 10.1021/ma002120x
-
[18]
Naoko, K., Yasuhiko, Y., Yoshio, I., Chûjô, R. and Yoshiharu, D., Macromolecules, 1989, 22:1676 doi: 10.1021/ma00194a030
-
[19]
Naoko, Y., Hiroyuki, M., Hidenori, S. and Yoshio, I., Macromolecules, 1995, 28:6516 doi: 10.1021/ma00123a018
-
[20]
Mitomo, H., Barham, P.J. and Keller, A., Polym. J., 1987, 19:1241 doi: 10.1295/polymj.19.1241
-
[21]
Mitomo, H., Barham, P.J. and Keller, A., Seni-Gakkaishi, 1986, 42:T589 doi: 10.2115/fiber.42.11_T589
-
[22]
Organ, S.J. and Barham, P.J., J. Mater. Sci., 1991, 26:1368 doi: 10.1007/BF00544479
-
[23]
Rule, R.J. and Liggat, J.J., Polymer, 1995, 36:3831 doi: 10.1016/0032-3861(95)99777-R
-
[24]
Holmes, P.A., Phys. Technol., 1985, 16:32 doi: 10.1088/0305-4624/16/1/305
-
[25]
Doi, Y., Tamaki, A., Kunioka, M. and Soga, K., Appl. Microbiol. Biotechnol., 1988, 28:330 doi: 10.1007/BF00268190
-
[26]
Vonk, C.G., Kortleve, G. and Koll, Z.Z., Polymer, 1967, 220:19 doi: 10.1007/BF02086052
-
[27]
Vonk, C.G., J. Appl. Cryst., 1971, 4:340 doi: 10.1107/S0021889871007179
-
[28]
Strobl, G.R. and Schneider, M.J., J. Polym. Sci., Polym. Phys. Ed., 1980, 18:1343 doi: 10.1002/pol.1980.180180614
-
[29]
Inoue, Y., Kamiya, N., Yamamoto, Y., Chûjô, R. and Yoshiharu, D., Macromolecules, 1989, 22:3800 doi: 10.1021/ma00199a051
-
[30]
Cuesta, M.S., Salazar, J.M., Barker, P.A. and Barham, P.J., J. Mater. Sci., 1992, 27:5335 doi: 10.1007/BF02403840
-
[1]
-

 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 796
- HTML全文浏览量: 15
 下载:
下载:
