 图 1
Mg-Co复合氧化物的XRD谱图
Figure 1.
XRD patterns of Mg-Co composite oxides with different compositions
图 1
Mg-Co复合氧化物的XRD谱图
Figure 1.
XRD patterns of Mg-Co composite oxides with different compositions
引用本文:
郑丽, 吴藏藏, 徐秀峰. N2O在Mg-Co和Mg-Mn-Co复合氧化物上的催化分解[J]. 燃料化学学报,
2016, 44(12): 1494-1501.

Citation: ZHENG Li, WU Cang-cang, XU Xiu-feng. Catalytic decomposition of N2O over Mg-Co and Mg-Mn-Co composite oxides[J]. Journal of Fuel Chemistry and Technology, 2016, 44(12): 1494-1501.
Citation: ZHENG Li, WU Cang-cang, XU Xiu-feng. Catalytic decomposition of N2O over Mg-Co and Mg-Mn-Co composite oxides[J]. Journal of Fuel Chemistry and Technology, 2016, 44(12): 1494-1501.
N2O在Mg-Co和Mg-Mn-Co复合氧化物上的催化分解
摘要:
用溶胶-凝胶法制备了不同组成的Mg-Co和Mg-Mn-Co复合氧化物,用于催化分解N2O。在较高活性的Mg-Mn-Co表面浸渍K2CO3溶液,制备K改性催化剂。用X射线衍射(XRD)、N2物理吸附(BET)、扫描电镜(SEM)、H2程序升温还原(H2-TPR)、O2程序升温脱附(O2-TPD)等技术表征催化剂结构,考察了复合氧化物的组成、K负载量等制备参数对催化剂活性的影响。结果表明,加入助剂K显著提高了催化剂活性,其中,0.02 K/MgMn0.2Co1.8O4活性较高,有氧无水、有氧有水气氛400℃连续反应50 h,N2O转化率分别保持97%和60%。有水-无水气氛交替实验表明,有水反应后再进行无水实验,K改性催化剂的稳定性较好。
-
关键词:
- N2O催化分解
- / Mg-Co复合氧化物
- / Mg-Mn-Co复合氧化物
- / K改性催化剂
- / 催化性能
English
Catalytic decomposition of N2O over Mg-Co and Mg-Mn-Co composite oxides
Abstract:
Mg-Co and Mg-Mn-Co composite oxides with different compositions were prepared by sol-gel method for N2O catalytic decomposition in the presence of oxygen. Of Mg-Mn-Co catalysts, the one with higher activity was impregnated by K2CO3 solution to make K-modified catalyst. These catalysts were characterized by X-ray diffraction(XRD), nitrogen physisorption (BET), scanning electron microscopy(SEM), temperature-programmed reduction of hydrogen(H2-TPR), and temperature-programmed desorption of oxygen(O2-TPD). The effect of preparation parameters such as compositions and potassium loadings on their catalytic activity has been investigated. The results show that K-modified catalysts exhibit better activity and higher resistance towards water in contrast to un-modified catalyst due to the weakness of surface metal-oxygen bonds. Among these catalysts, 0.02K/MgMn0.2Co1.8O4 is the most active, over which 97% and 60% conversions of N2O can be reached at 400℃ after continuous running for 50 h under the atmosphere of oxygen-alone and oxygen-steam together, respectively. When the steam is switched off, the catalytic activity of 0.02K/MgMn0.2Co1.8O4 can be restored to large extent, indicating the good water-resistance of K-modified catalyst.
-
N2O是一种重要的温室气体,其增温潜能是CO2的310倍,寿命长达120年,还对大气臭氧层有严重的破坏作用。硝酸、己二酸生产是N2O的主要排放源。在催化剂作用下,把N2O分解成对环境无毒无害的N2和O2,是消除N2O的有效方法。文献[1-4]报道,N2O催化分解遵循氧化-还原机理,所以N2O分解催化剂必须含有可变价的金属离子。研究较多的催化剂有过渡金属氧化物[5-9]、负载型贵金属[10-13]、离子交换分子筛[14-16],其中,Co3O4尖晶石型氧化物是近年来研究较多的催化剂。为了提高催化剂活性,制备了钴系复合氧化物,用于催化分解N2O。Yan等[17, 18]用Mg2+、Zn2+分别取代Co3O4中的部分Co2+,制得MgxCo1-xCo2O4、ZnxCo1-xCo2O4复合氧化物。也有文献制备了两种金属离子均可变价的钴系复合氧化物,Abu-Zied等[19]在Co3O4中加入Ni组分,制备NixCo1-xCo2O4催化剂,Maniak等[20]用Fe取代Co3O4中的Co制得FexCo3-xO4催化剂。在前期工作中,本课题组[21, 22]用Cu2+、Mn2+分别取代Co3O4中部分Co,制得了CuxCo3-xO4、MnxCo3-xO4复合氧化物,用于催化分解N2O。文献[2, 3, 21, 22]报道,过渡金属氧化物表面负载少量碱金属助剂,可增加过渡金属粒子周围的电子云密度,降低其电子结合能,增强活性组分的还原能力和催化性能。
Mg-Co复合氧化物催化分解N2O的研究已有报道[23],催化剂多用共沉淀法制备。相比较而言,溶胶-凝胶法制备过程简单,催化剂组成易重复。本研究用溶胶-凝胶法制备了不同组成的Mg-Co复合氧化物,再引入锰元素制备了Mg-Mn-Co三元复合氧化物,表面浸渍K2CO3溶液,制备K改性催化剂,用于催化分解N2O,考察了催化剂组成和助剂用量对催化活性的影响,并考察了有水条件下催化剂的稳定性。
1 实验部分
1.1 催化剂的制备
1.2 N2O分解反应
N2O分解反应在固定式反应器中进行,有氧条件下反应气组成为2%N2O+4%O2+Ar(体积分数)。有氧有水条件下反应气组成为2%N2O+4%O2+8%H2O+Ar,总流量140mL/min。催化剂用量为1g,程序控温仪控制反应温度,反应尾气经六通阀进样,用GC-920型气相色谱仪(桥电流120mA,载气H2,固定相 Porapak Q,TCD检测器)检测N2O的剩余浓度。
催化剂的初活性测试: 程序升温,每个温度点恒温反应30min,测试N2O剩余浓度,计算N2O转化率。
催化剂的稳定性测试: 以10℃/min从室温程序升温到400℃,恒温反应50h,测定不同时刻N2O的剩余浓度,计算转化率。
1.3 催化剂的表征
1.1.2 Mg-Mn-Co复合氧化物
以Mg(NO3)2·6H2O、Co(NO3)2·6H2O、50%的Mn(NO3)2溶液为原料,配置总金属离子浓度为1mol/L的混合盐溶液,剧烈搅拌下缓慢滴加1mol/L柠檬酸溶液(柠檬酸与总金属离子的物质的量比为1∶1),用2.5%稀氨水调节溶液的pH值至2,搅拌30min,将母液在65℃旋转蒸发至透明胶状,水浴中静置2h,120℃干燥12h,600℃焙烧4h,制得Mg-Mn-Co系列催化剂,记为MgMnyCo2-yO4,其中,y取值0.2、0.4、0.6、0.8、1。
1.1.1 Mg-Co复合氧化物
按预定组成,配制金属离子总浓度为1mol/L的Mg(NO3)2和Co(NO3)2混合溶液,在25℃水浴中剧烈搅拌,逐滴加入1mol/L柠檬酸溶液(柠檬酸与总金属离子的物质的量比控制在1∶1),并滴加2.5%(质量分数)稀氨水调节pH值至2,继续搅拌30min。将上述溶液在65℃水浴中旋转蒸发至透明胶状,65℃水浴中静置2h,然后120℃干燥12h,600℃焙烧4h,得到MgxCo3-xO4(x=0.2、0.4、0.6、0.8、1)系列的催化剂。
1.1.3 K改性MgMn0.2Co1.8O4
先制备MgMn0.2Co1.8O4催化剂。按不同K/(Mn+Co)物质的量比,配制要求浓度的K2CO3溶液,在室温下等体积浸渍MgMn0.2Co1.8O4样品各24h,120℃干燥12h,600℃焙烧4h,制得K改性催化剂,记为zK/MgMn0.2Co1.8O4,其中,z为K/(Mn+Co)物质的量比。
1.3.4 O2程序升温脱附
测试仪器为北京彼奥德公司生产的PCA-1200型化学吸附仪,样品用量约100mg。测试前对样品进行预处理:在O2中从室温升至120℃(升温速率10℃/min),恒温吸附30min,然后冷却至室温。关闭O2,打开He,待基线平稳后,程序升温至900℃(升温速率10℃/min),TCD检测器记录脱氧信息。
1.3.5 形貌分析
测试仪器为日本Hitachi公司生产的S-4800型扫描电镜。为增加样品的导电性,测试前用E-1045型离子溅射仪(Hitachi公司生产)对样品表面进行喷Pt处理。
1.3.2 比表面积测定
测试仪器为美国Quantachrome公司生产的NOVA3000型自动吸附仪,测试前样品先经300℃减压处理2h,除去表面吸附的杂质。N2为吸附气,液氮温度下吸附,室温脱附,用BET公式计算催化剂的比表面积。
1.3.1 物相分析
测试仪器为日本岛津XRD-6100型X射线衍射仪,Cu Kα射线,石墨单色器,管电压、管电流分别为40kV和30mA。闪烁计数器记录衍射强度。根据尖晶石(311)晶面的衍射数据,用Scherrer方程计算催化剂的晶粒粒径:
式中,K为Scherrer常数,取值0.89;β为(311)晶面衍射峰的半高宽度,θ是(311)晶面对应的衍射角。
1.3.3 H2程序升温还原
测试仪器为北京彼奥德公司生产的PCA-1200型化学吸附仪,样品用量约80mg。测试前对样品进行预处理:在Ar中从室温升至500℃(升温速率10℃/min),恒温吹扫30min,然后冷却至室温。关闭Ar,打开10% H2/Ar还原气,流量为20mL/min,程序升温至900℃(升温速率10℃/min),TCD检测器记录耗氢信息。
2 结果与讨论
2.1 MgxCo3-xO4复合氧化物的结构表征与催化活性
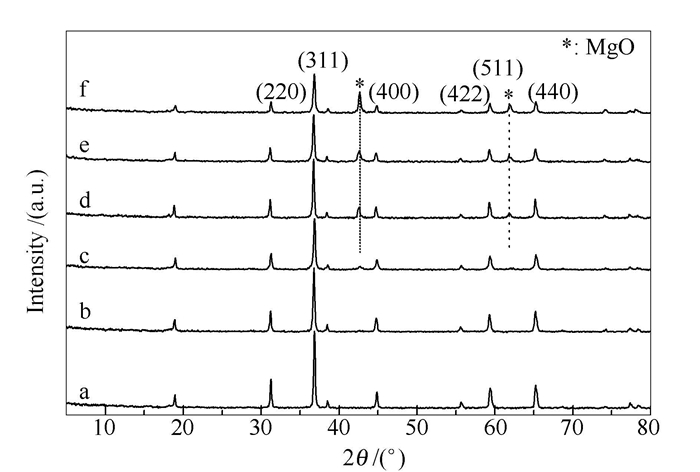
图 1为不同组成Mg-Co复合氧化物的XRD谱图,由图 1可知,各催化剂均呈现出尖晶石 (220)、(311)、(440)、(511)等晶面的特征衍射峰,说明Mg-Co复合氧化物的晶相结构均为尖晶石型。但随着Mg含量的增多,Mg0.4Co2.6O4、Mg0.6Co2.4O4、Mg0.8Co2.2O4、MgCo2O4中出现了MgO杂相,这是因为催化剂制备时加入了柠檬酸,焙烧过程中有机胶凝剂的还原作用致使低价态Co2+增多,按尖晶石的AB2O4型结构,部分二价离子(Mg2+)不能进入尖晶石晶格而成为自由相。
图 1
Mg-Co复合氧化物的XRD谱图
Figure 1.
XRD patterns of Mg-Co composite oxides with different compositions
图 2为Mg-Co复合氧化物上N2O转化率数据,由图 2可以看出,加入适量Mg提高了催化剂活性,其中,MgCo2O4活性较高,这与其较大的比表面积和较小的晶粒粒径相关(见表 1)。文献[24, 25]报道:N2O分解过程中,N2O首先吸附在催化剂表面活性位(*)上,裂解生成吸附态氧原子O*和产物N2,活性氧原子O*相互反应生成O2,从催化剂表面脱附下来(慢步骤),所以活性较高的催化剂应具有较好的氧脱除能力。
 图 2
Mg-Co复合氧化物上的N2O转化率
Figure 2.
N2O conversions over Mg-Co composite oxides with different compositions
图 2
Mg-Co复合氧化物上的N2O转化率
Figure 2.
N2O conversions over Mg-Co composite oxides with different compositions
 表 1
Mg-Co复合氧化物的晶粒粒径和比表面积
Table 1.
Crystallite size and BET surface area of Mg-Co composite oxides
表 1
Mg-Co复合氧化物的晶粒粒径和比表面积
Table 1.
Crystallite size and BET surface area of Mg-Co composite oxides
d/nma
A/(m2·g-1)Mg0.2Co2.8O4 85.0 8.4 Mg0.4Co2.6O4 66.1 13.0 Mg0.6Co2.4O4 104.4 18.8 Mg0.8Co2.2O4 74.1 16.8 acalculated by Scherrer equation on the basis of (311) crystallographic plane data in XRD patterns 表 1 Mg-Co复合氧化物的晶粒粒径和比表面积
Table 1. Crystallite size and BET surface area of Mg-Co composite oxides图 3为催化剂的H2-TPR谱图。由图 3可知,Co3O4的H2-TPR谱图中,250-400→℃的低温还原峰、400-600→℃的高温还原峰分别归属于Co3+→Co2+和Co2+→Co0。根据Co3O4的组成,Co3+→Co2+与Co2+→Co0的理论耗氢量之比应为1∶3,这与图 3a谱线的两个峰面积之比基本一致[26]。与Co3O4相比,Mg-Co复合催化剂的低温还原峰(Co3+→Co2+)向低温区偏移,而高温还原峰(Co2+→Co0)发生了拖尾(结束温度有所升高)。相比较而言,MgCo2O4的Co3+→Co2+还原温度低于其他Mg-Co催化剂,说明其还原(氧脱除)能力较强,因而有较高的催化活性。
 图 3
Mg-Co复合氧化物的H2-TPR谱图
Figure 3.
H2-TPR profiles of Mg-Co composite oxides with different compositions
图 3
Mg-Co复合氧化物的H2-TPR谱图
Figure 3.
H2-TPR profiles of Mg-Co composite oxides with different compositions
图 4为Mg-Co复合氧化物的O2-TPD谱图,由图 4可知,与Mg0.2Co2.8O4相比,Mg0.4Co2.6O4、Mg0.6Co2.4O4、Mg0.8Co2.2O4、MgCo2O4的氧脱附峰向低温区偏移,其中,MgCo2O4的氧脱附量较大,说明其表面吸氧量多、与氧物种的相互作用又较弱(吸附氧易脱除),这与MgCo2O4较高的催化活性正相关。
 图 4
Mg-Co复合氧化物的O2-TPD谱图
Figure 4.
O2-TPD profiles of Mg-Co composite oxides with different compositions
图 4
Mg-Co复合氧化物的O2-TPD谱图
Figure 4.
O2-TPD profiles of Mg-Co composite oxides with different compositions
2.2 MgMnyCo2-yO4复合氧化物的结构表征与催化活性
总结以上结果,组成为MgCo2O4的催化剂活性较高。为了进一步提高催化活性,用Mn取代MgCo2O4中部分Co元素,制备了MgMnyCo2-yO4催化剂。
图 5为Mg-Mn-Co复合氧化物的XRD谱图。由图 5可知,除了尖晶石物相,Mn含量较低的MgCo2O4、MgMn0.2Co1.8O4、MgMn0.4Co1.6O4还出现了MgO杂相。随着Mn含量的增多,MgO物相峰减弱,MgMn0.6Co1.4O4、MgMn0.8Co1.2O4、MgMnCoO4中MgO物相已完全消失,这可能因为锰离子比钴离子更易呈现高价态,使得二价Mg2+离子全部进入了尖晶石晶格。
 图 5
Mg-Mn-Co复合氧化物的XRD谱图
Figure 5.
XRD patterns of Mg-Mn-Co composite oxides with different compositions
图 5
Mg-Mn-Co复合氧化物的XRD谱图
Figure 5.
XRD patterns of Mg-Mn-Co composite oxides with different compositions
表 2为Mg-Mn-Co复合氧化物的晶粒粒径和比表面积测试数据,与MgCo2O4相比,三元催化剂的晶粒粒径有所减小、比表面积增大。SEM照片(图 6)也表明,Mg-Mn-Co催化剂颗粒粒径明显小于MgCo2O4。
表 2
Mg-Mn-Co复合氧化物的晶粒粒径和比表面积
Table 2.
Crystallite size and BET Surface area of Mg-Mn-Co composite oxides
d/nma
A/(m2·g-1)MgMn0.2Co1.8O4 17.6 75.0 MgMn0.4Co1.6O4 7.8 77.7 MgMn0.6Co1.4O4 8.6 81.0 MgMn0.8Co1.2O4 16.0 72.4 a calculated by Scherrer equation on the basis of (311) crystallographic plane data in XRD patterns 表 2 Mg-Mn-Co复合氧化物的晶粒粒径和比表面积
Table 2. Crystallite size and BET Surface area of Mg-Mn-Co composite oxides 图 6
Mg-Mn-Co复合氧化物的SEM照片
Figure 6.
SEM images of Mg-Mn-Co composite oxides with different compositions
图 6
Mg-Mn-Co复合氧化物的SEM照片
Figure 6.
SEM images of Mg-Mn-Co composite oxides with different compositions
图 7为Mg-Mn-Co复合氧化物上的N2O转化率。由图 7可知,适量Mn的加入提高了催化剂活性,其中,MgMn0.2Co1.8O4的催化活性最高。
 图 7
Mg-Mn-Co复合氧化物的催化活性
Figure 7.
N2O conversions over Mg-Mn-Co composite oxides with different compositions
图 7
Mg-Mn-Co复合氧化物的催化活性
Figure 7.
N2O conversions over Mg-Mn-Co composite oxides with different compositions
图 8为Mg-Mn-Co复合氧化物的H2-TPR谱图。由图 8可知,MgCo2O4催化剂中,250-390℃的低温还原峰归属于Co3+→Co2+,400-600℃的高温还原峰归属于Co2+→Co0。对于Mg-Mn-Co复合氧化物,其低温还原峰(250-450℃)归属于Co3+→Co2+和Mn2O3(或Mn3O4)→MnO,峰型比Mg-Co催化剂宽的多,应与MnOx的还原有关。研究对比可以发现,MgMn0.2Co1.8O4的低温还原峰温比MgCo2O4明显前移,MgMn0.4Co1.6O4和MgMn0.6Co1.4O4的低温还原峰温与MgCo2O4相近,而MgMn0.8Co1.2O4、MgMnCoO4的低温还原峰温又发生了后移。根据以上数据,可以认为MgMn0.2Co1.8O4较高的催化活性与其较易还原(脱除氧)的性质有关。
 图 8
Mg-Mn-Co复合氧化物的H2-TPR谱图
Figure 8.
H2-TPR profiles of Mg-Mn-Co composite oxides with different compositions
图 8
Mg-Mn-Co复合氧化物的H2-TPR谱图
Figure 8.
H2-TPR profiles of Mg-Mn-Co composite oxides with different compositions
图 9为Mg-Mn-Co复合氧化物的O2-TPD谱图。为了与催化剂的反应温度相对应,我们截取了100-500℃的O2-TPD谱图进行研究。将较低(低于200℃)和较高温度(300-350℃)区间的脱附氧分别归属为催化剂表面弱、强吸附的氧物种[27, 28]。与MgCo2O4相比,Mg-Mn-Co复合氧化物的低温脱附峰向低温区偏移,说明其表面氧易脱除。对比不同组成的Mg-Mn-Co复合氧化物,MgMn0.2Co1.8O4的表面氧易脱除、且氧脱附量较大,有较高的催化活性。
 图 9
Mg-Mn-Co复合氧化物的O2-TPD谱图
Figure 9.
O2-TPD profiles of Mg-Mn-Co composite oxides with different compositions
图 9
Mg-Mn-Co复合氧化物的O2-TPD谱图
Figure 9.
O2-TPD profiles of Mg-Mn-Co composite oxides with different compositions
2.3 K改性Mg-Mn-Co催化剂的结构表征与催化活性
综合以上结果,组成为MgMn0.2Co1.8O4的催化剂具有较高的催化活性。为了进一步提高催化活性,在其表面浸渍K2CO3溶液,制备K改性催化剂。
XRD测试结果表明,载K催化剂均为尖晶石晶型。表 3为催化剂的晶粒粒径和比表面积数据,与MgMn0.2Co1.8O4(75.0m2/g)相比,K改性催化剂的晶粒粒径较大、比表面积有所减小。SEM照片(图 10)表明,0.02K/MgMn0.2Co1.8O4催化剂呈片状,颗粒粒径明显大于未改性催化剂。
表 3
K/MgMn0.2Co1.8O4催化剂的晶粒粒径和比表面积
Table 3.
Crystallite size and BET surface area of K/MgMn0.2Co1.8O4 catalysts
d/nma
A/(m2·g-1)0.01K/MgMn0.2Co1.8O4 21.2 60.8 0.02K/MgMn0.2Co1.8O4 33.6 54.6 0.03K/MgMn0.2Co1.8O4 25.4 45.7 0.04K/MgMn0.2Co1.8O4 24.7 56.8 a calculated by Scherrer equation on the basis of (311) crystallographic plane data in XRD patterns 表 3 K/MgMn0.2Co1.8O4催化剂的晶粒粒径和比表面积
Table 3. Crystallite size and BET surface area of K/MgMn0.2Co1.8O4 catalysts 图 10
K/MgMn0.2Co1.8O4催化剂的SEM照片
Figure 10.
SEM images of K/MgMn0.2Co1.8O4 catalysts
图 10
K/MgMn0.2Co1.8O4催化剂的SEM照片
Figure 10.
SEM images of K/MgMn0.2Co1.8O4 catalysts
图 11为K改性催化剂上的N2O转化率。由图 11可知,K改性催化剂上N2O在450℃时全分解,K改性催化剂活性显著优于未改性催化剂。尤其0.02K/MgMn0.2Co1.8O4的催化活性更高,375℃反应N2O已完全转化。
 图 11
K/MgMn0.2Co1.8O4催化剂的催化活性
Figure 11.
N2O conversions over K/MgMn0.2Co1.8O4 catalysts
图 11
K/MgMn0.2Co1.8O4催化剂的催化活性
Figure 11.
N2O conversions over K/MgMn0.2Co1.8O4 catalysts
图 12为K改性催化剂的H2-TPR谱图,各催化剂在250-400℃的低温还原峰归属于Co3+→Co2+和Mn2O3(或Mn3O4)→MnO,400-800℃的高温还原峰归属于Co2+→Co0。与MgMn0.2Co1.8O4相比,K改性催化剂的低温还原峰向低温偏移,说明其还原反应容易进行,氧脱除能力较强,因而有较高的催化活性。
 图 12
K/MgMn0.2Co1.8O4催化剂的H2-TPR谱图
Figure 12.
H2-TPR profiles of K/MgMn0.2Co1.8O4 catalysts
图 12
K/MgMn0.2Co1.8O4催化剂的H2-TPR谱图
Figure 12.
H2-TPR profiles of K/MgMn0.2Co1.8O4 catalysts
2.4 有氧有水条件下0.02K/MgMn0.2Co1.8O4催化剂的稳定性
以上实验在有氧条件下优化出了活性较高的0.02K/MgMn0.2Co1.8O4催化剂,下面在有氧有水条件下测试催化剂的稳定性。图 13为不同气氛中0.02K/MgMn0.2Co1.8O4催化剂的稳定性。
 图 13
不同气氛中0.02K/MgMn0.2Co1.8O4
催化剂的稳定性
Figure 13.
Catalytic stability of 0.02 K/MgMn0.2Co1.8O4
for N2O decomposition at 400℃ under various atmospheres
图 13
不同气氛中0.02K/MgMn0.2Co1.8O4
催化剂的稳定性
Figure 13.
Catalytic stability of 0.02 K/MgMn0.2Co1.8O4
for N2O decomposition at 400℃ under various atmospheres
由图 13可知,有氧条件下400℃连续反应50h,未改性和K改性催化剂均有较好的稳定性,N2O转化率分别保持64%、97%。而有氧有水条件下,400℃连续反应50h,未改性和K改性催化剂上的N2O转化率分别降至23%、60%,水汽的存在明显降低了催化剂的活性,这可能因为水在催化剂表面活性位上优先吸附,阻止了N2O的吸附活化。相比较而言,K改性催化剂优于未改性催化剂。
对0.02K/MgMn0.2Co1.8O4催化剂进行了有水-无水交替实验,具体见图 14。由图 14可知,发现有水反应后再进行无水实验,N2O转化率有所下降,但下降幅度不大,催化剂稳定性较好。
 图 14
0.02K/MgMn0.2Co1.8O4催化剂无水-有水交替反应的催化活性
Figure 14.
Catalytic activity of 0.02K/MgMn0.2Co1.8O4
for N2O decomposition at 400℃ under oxygen-only or
oxygen-steam atmospheres
图 14
0.02K/MgMn0.2Co1.8O4催化剂无水-有水交替反应的催化活性
Figure 14.
Catalytic activity of 0.02K/MgMn0.2Co1.8O4
for N2O decomposition at 400℃ under oxygen-only or
oxygen-steam atmospheres
3 结 论
用Mg取代Co3O4中部分Co制备的Mg-Co复合氧化物,提高了催化剂的比表面积和催化活性,其中,组成为MgCo2O4的催化剂活性较高。
在MgCo2O4基础上,用Mn取代部分Co制备Mg-Mn-Co三元复合氧化物,优化出了活性较高的MgMn0.2Co1.8O4催化剂。
在MgMn0.2Co1.8O4表面浸渍K2CO3溶液,制备了K改性催化剂。活性较高的催化剂是0.02K/MgMn0.2Co1.8O4,有氧无水、有氧有水条件下400℃连续反应50h,N2O转化率分别保持97%和60%。有水-无水交替实验表明,有水反应后再进行无水反应,0.02K/MgMn0.2Co1.8O4催化剂的稳定性较好。
-
-
[1]
PIETROGIACOMI D, CAMPA M C, CARBONE L R, TUTI S, OCCHIUZZI M. N2O decomposition on CoOx,CuOx,FeOx or MnOx supported on ZrO2:The effect of zirconia doping with sulfates or K+ on catalytic activity[J]. Appl Catal B:Environ, 2016, 187: 218-227. doi: 10.1016/j.apcatb.2016.01.018
-
[2]
ASANO K, OHNISHI C, IWAMOTO S, SHIOYA Y, INOUE M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O[J]. Appl Catal B:Environ, 2008, 78(3/4): 242-249.
-
[3]
XUE L, ZHANG C B, HE H, TERAOKA Y. Promotion effect of residual K on the decomposition of N2O over cobalt-cerium mixed oxide catalyst[J]. Catal Today, 2007, 126(3/4): 449-455.
-
[4]
HUSSAIN M, FINO D, RUSSO N. N2O decomposition by mesoporous silica supported Rh catalysts[J]. J Hazard Mater, 2012, 211-212: 255-265. doi: 10.1016/j.jhazmat.2011.08.024
-
[5]
KLYUSHINA, PACULTOVÁK, KREJČOVÁS, SLOWIK, JIRÁTOVÁK, KOVANDA, RYCZKOWSKI, OBALOVÁL. Advantages of stainless steel sieves as support for catalytic N2O decomposition over K-doped Co3O4[J]. Catal Today, 2015, 257(1): 2-10.
-
[6]
ZABILSKIY M, DJINOVĆ P, ERJAVEC B, DRAŽĆG , PINTAR A. Small CuO clusters on CeO2 nanospheres as active species for catalytic N2O decomposition[J]. Appl Catal B:Environ, 2015, 163: 113-122. doi: 10.1016/j.apcatb.2014.07.057
-
[7]
AMROUSSE R, KATSUMI T. Substituted ferrite MxFe1-xFe2O4(M=Mn,Zn) catalysts for N2O catalytic decomposition processes[J]. Catal Commun, 2012, 26: 194-198. doi: 10.1016/j.catcom.2012.05.024
-
[8]
KUMAR S, VINU A, SUBBRT J, BAKARDJIEVA S, RAYALU S, TERAOKA Y, LABHSETWAR N. Catalytic N2O decomposition on Pr0.8Ba0.2MnO3 type perovskite catalyst for industrial emission control[J]. Catal Today, 2012, 198(1): 125-132. doi: 10.1016/j.cattod.2012.06.015
-
[9]
XUE Z W, SHEN Y S, SHEN S B, LI C L, ZHU S M. Promotional effects of Ce4+,La3+ and Nd3+ incorporations on catalytic performance of Cu-Fe-Ox for decomposition of N2O[J]. J Ind Eng Chem, 2015, 30: 98-105. doi: 10.1016/j.jiec.2015.05.008
-
[10]
LIN Y, MENG T, MA Z. Catalytic decomposition of N2O over RhOx supported on metal phosphates[J]. J Ind Eng Chem, 2015, 28: 138-146. doi: 10.1016/j.jiec.2015.02.009
-
[11]
DACQUIN J P, DUJARDIN C, GRANGER P. Surface reconstruction of supported Pd on LaCoO3:Consequences on the catalytic properties in the decomposition of N2O[J]. J Catal, 2008, 253(1): 37-49. doi: 10.1016/j.jcat.2007.10.023
-
[12]
BEYER H, EMMERICH J, CHATZIAPOSTOLOU K, KÖHLER K. Decomposition of nitrous oxide by rhodium catalysts:Effect of rhodium particle size and metal oxide support[J]. Appl Catal A:Gen, 2011, 391(1/2): 411-416.
-
[13]
PACHATOURIDOU E, PAPISTA E, DELIMITIS A, VASILIADES M A, EFSTATHIOU A M, AMIRIDIS M D, ALEXEEV O S, BLOOM D, MAMELLOS G E, KONSOLAKIS M, ILIOPOULOU E. N2O decomposition over ceria-promoted Ir/Al2O3 catalysts:The role of ceria[J]. Appl Catal B:Environ, 2016, 187: 259-268. doi: 10.1016/j.apcatb.2016.01.049
-
[14]
BERRIER E, OVSITSER O, KONDRATENKO E V, SCHWIDDER M, GRÜNERT W, BRÜCKNER A. Temperature-dependent N2O decomposition over Fe-ZSM-5:Identification of sites with different activity[J]. J Catal, 2007, 249(1): 67-78. doi: 10.1016/j.jcat.2007.03.027
-
[15]
CÜRDANELI P E, ÖZKAR S. Ruthenium (Ⅲ) ion-exchanged zeolite Y as highly active and reusable catalyst in decomposition of nitrous oxide to sole nitrogen and oxygen[J]. Microporous Mesoporous Mater, 2014, 196: 51-58. doi: 10.1016/j.micromeso.2014.04.052
-
[16]
MENG T, REN N, MA Z. Silicalite-1@Cu-ZSM-5 core-shell catalyst for N2O decomposition[J]. J Mol Catal A:Chem, 2015, 404-405: 233-239. doi: 10.1016/j.molcata.2015.05.006
-
[17]
YAN L, REN T, WANG X L, GAO Q, JI D, SUO J S. Excellent catalytic performance of ZnxCo1-xCo2O4 spinel catalysts for the decomposition of nitrous oxide[J]. Catal Commun, 2003, 4(10): 505-509. doi: 10.1016/S1566-7367(03)00131-6
-
[18]
YAN L, REN T, WANG X L, JI D, SUO J S. Catalytic decomposition of N2O over MxCo1-xCo2O4(M=Ni,Mg) spinel oxides[J]. Appl Catal B:Environ, 2003, 45(2): 85-90. doi: 10.1016/S0926-3373(03)00174-7
-
[19]
ABU-ZIED B M, SOLIMAN S A, ABDELLAH S E. Pure and Ni-substituted Co3O4 spinel catalysts for direct N2O decomposition[J]. Chin J Catal, 2014, 35(7): 1105-1112. doi: 10.1016/S1872-2067(14)60058-9
-
[20]
MANIAK G, STELMACHOWSKI P, STANEK J J, KOTARBA A, SOJKA Z. Catalytic properties in N2O decomposition of mixed cobalt-iron spinels[J]. Catal Commun, 2011, 15(1): 127-131. doi: 10.1016/j.catcom.2011.08.027
-
[21]
窦喆, 张海杰, 潘燕飞, 徐秀峰. N2O在钾改性Cu-Co尖晶石型复合氧化物上的催化分解[J]. 燃料化学学报, 2014,42,(2): 238-245. doi: 10.1016/S1872-5813(14)60016-5DOU Zhe, ZHANG Hai-jie, PAN Yan-fei, XU Xiu-feng. Catalytic decomposition of N2O over potassium-modified Cu-Co spinel oxides[J]. J Fuel Chem Technol, 2014, 42(2): 238-245. doi: 10.1016/S1872-5813(14)60016-5
-
[22]
王建, 窦喆, 潘燕飞, 徐秀峰. MnxCo3-xO4复合氧化物及改性催化剂催化分解N2O[J]. 分子催化, 2015,29,(3): 246-255. WANG Jian, DOU Zhe, PAN Yan-fei, XU Xiu-feng. MnxCo3-xO4 composite oxide and modified catalyst catalytic decomposition of N2O[J]. J Mol Catal (China), 2015, 29(3): 246-255.
-
[23]
CHELLAM U, XU Z P, ZENG H C. Low-temperature synthesis of MgxCo1-xCo2O4 spinel catalysts for N2O decomposition[J]. Chem Mater, 2000, 12: 650-658. doi: 10.1021/cm990355l
-
[24]
KUBOŇOVÁL , FRIDRICHOVÁD , WACH A, KUŚTROWSKR P, OBALOVÁL , COOL P. Catalytic activity of rhodium grafted on ordered mesoporous silicamaterials modified with aluminum in N2O decomposition[J]. Catal Today, 2015, 257: 51-58. doi: 10.1016/j.cattod.2015.03.019
-
[25]
PRRUTKO L V, CHERNYAVSKY V S, STAROKON E V, IVANOV A A, KHARITONOV A S, PANOV G. The role of a-sites in N2O decomposition over FeZSM-5.Comparison with the oxidation of benzene to phenol[J]. Appl Catal B:Environ, 2009, 91(1/2): 174-179.
-
[26]
吴藏藏, 张海杰, 王建, 郑丽, 徐秀峰. N2O分解催化剂Co-Al尖晶石型复合氧化物制备参数的优化[J]. 分子催化, 2016,30,(1): 62-71. WU Cang-cang, ZHANG Hai-jie, WANG Jian, ZHENG Li, XU Xiu-feng. The preparation parameters screening of Co-Al spinel oxides for N2O catalytic decomposition[J]. J Mol Catal (China), 2016, 30(1): 62-71.
-
[27]
AMROUSSE R, TSUTSUMI A, BACHAR A, LAHCENE D. N2O catalytic decomposition over nano-sized particles of Co-substituted Fe3O4 substrates[J]. Appl Catal A:Gen, 2013, 450: 253-260. doi: 10.1016/j.apcata.2012.10.036
-
[28]
FRANKEN T, PALKOVITS R. Investigation of potassium doped mixed spinels CuxCo3-xO4 as catalysts for an efficient N2O decomposition in real reaction conditions[J]. Appl Catal B:Environ, 2015, 176-177: 298-305. doi: 10.1016/j.apcatb.2015.04.002
-
[1]
-

图 1 Mg-Co复合氧化物的XRD谱图
Figure 1 XRD patterns of Mg-Co composite oxides with different compositions
a: Co3O4; b: Mg0.2Co2.8O4; c: Mg0.4Co2.6O4; d: Mg0.6Co2.4O4; e: Mg0.8Co2.2O4; f: MgCo2O4

图 2 Mg-Co复合氧化物上的N2O转化率
Figure 2 N2O conversions over Mg-Co composite oxides with different compositions

图 3 Mg-Co复合氧化物的H2-TPR谱图
Figure 3 H2-TPR profiles of Mg-Co composite oxides with different compositions
a: Co3O4; b: Mg0.2Co2.8O4; c: Mg0.4Co2.6O4; d: Mg0.6Co2.4O4; e: Mg0.8Co2.2O4; f: MgCo2O4

图 4 Mg-Co复合氧化物的O2-TPD谱图
Figure 4 O2-TPD profiles of Mg-Co composite oxides with different compositions
a: Mg0.2Co2.8O4; b: Mg0.4Co2.6O4; c: Mg0.6Co2.4O4; d: Mg0.8Co2.2O4; e: MgCo2O4

图 5 Mg-Mn-Co复合氧化物的XRD谱图
Figure 5 XRD patterns of Mg-Mn-Co composite oxides with different compositions
a: MgCo2O4; b: MgMn0.2Co1.8O4; c: MgMn0.4Co1.6O4; d: MgMn0.6Co1.4O4; e: MgMn0.8Co1.2O4; f: MgMnCoO4

图 6 Mg-Mn-Co复合氧化物的SEM照片
Figure 6 SEM images of Mg-Mn-Co composite oxides with different compositions
(a): MgCo2O4; (b): MgMn0.2Co1.8O4; (c): MgMn0.6Co1.4O4; (d): MgMnCoO4

图 7 Mg-Mn-Co复合氧化物的催化活性
Figure 7 N2O conversions over Mg-Mn-Co composite oxides with different compositions

图 8 Mg-Mn-Co复合氧化物的H2-TPR谱图
Figure 8 H2-TPR profiles of Mg-Mn-Co composite oxides with different compositions
a: MgCo2O4; b: MgMn0.2Co1.8O4; c: MgMn0.4Co1.6O4; d: MgMn0.6Co1.4O4; e: MgMn0.8Co1.2O 4; f: MgMnCoO4

图 9 Mg-Mn-Co复合氧化物的O2-TPD谱图
Figure 9 O2-TPD profiles of Mg-Mn-Co composite oxides with different compositions
a: MgCo2O4; b: MgMn0.2Co1.8O4; c: MgMn0.4Co1.6O4; d: MgMn0.6Co1.4O4; e: MgMn0.8Co1.2O 4; f: MgMnCoO4

图 10 K/MgMn0.2Co1.8O4催化剂的SEM照片
Figure 10 SEM images of K/MgMn0.2Co1.8O4 catalysts
(a): MgMn0.2Co1.8O4; (b): 0.02K/MgMn0.2Co1.8O4

图 11 K/MgMn0.2Co1.8O4催化剂的催化活性
Figure 11 N2O conversions over K/MgMn0.2Co1.8O4 catalysts

图 12 K/MgMn0.2Co1.8O4催化剂的H2-TPR谱图
Figure 12 H2-TPR profiles of K/MgMn0.2Co1.8O4 catalysts
a: MgMn0.2Co1.8O4; b: 0.01K/MgMn0.2Co1.8O4; c: 0.02K/MgMn0.2Co1.8O4; d: 0.03K/MgMn0.2Co1.8O4; e: 0.04K/MgMn0.2Co1.8O4; f: 0.05K/MgMn0.2Co1.8O4

图 13 不同气氛中0.02K/MgMn0.2Co1.8O4 催化剂的稳定性
Figure 13 Catalytic stability of 0.02 K/MgMn0.2Co1.8O4 for N2O decomposition at 400℃ under various atmospheres
■: MgMn0.2Co1.8O4 (O2); ●:MgMn0.2Co1.8O4(O2+H2O);▲: 0.02K/MgMn0.2Co1.8O4(O2); ▼: 0.02K/MgMn0.2Co1.8O4(O2+H2O)

图 14 0.02K/MgMn0.2Co1.8O4催化剂无水-有水交替反应的催化活性
Figure 14 Catalytic activity of 0.02K/MgMn0.2Co1.8O4 for N2O decomposition at 400℃ under oxygen-only or oxygen-steam atmospheres
表 1 Mg-Co复合氧化物的晶粒粒径和比表面积
Table 1. Crystallite size and BET surface area of Mg-Co composite oxides
d/nma
A/(m2·g-1)Mg0.2Co2.8O4 85.0 8.4 Mg0.4Co2.6O4 66.1 13.0 Mg0.6Co2.4O4 104.4 18.8 Mg0.8Co2.2O4 74.1 16.8 acalculated by Scherrer equation on the basis of (311) crystallographic plane data in XRD patterns  下载: 导出CSV
下载: 导出CSV
表 2 Mg-Mn-Co复合氧化物的晶粒粒径和比表面积
Table 2. Crystallite size and BET Surface area of Mg-Mn-Co composite oxides
d/nma
A/(m2·g-1)MgMn0.2Co1.8O4 17.6 75.0 MgMn0.4Co1.6O4 7.8 77.7 MgMn0.6Co1.4O4 8.6 81.0 MgMn0.8Co1.2O4 16.0 72.4 a calculated by Scherrer equation on the basis of (311) crystallographic plane data in XRD patterns
下载: 导出CSV
表 3 K/MgMn0.2Co1.8O4催化剂的晶粒粒径和比表面积
Table 3. Crystallite size and BET surface area of K/MgMn0.2Co1.8O4 catalysts
d/nma
A/(m2·g-1)0.01K/MgMn0.2Co1.8O4 21.2 60.8 0.02K/MgMn0.2Co1.8O4 33.6 54.6 0.03K/MgMn0.2Co1.8O4 25.4 45.7 0.04K/MgMn0.2Co1.8O4 24.7 56.8 a calculated by Scherrer equation on the basis of (311) crystallographic plane data in XRD patterns
下载: 导出CSV
-















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1
- 文章访问数: 943
- HTML全文浏览量: 208
 下载:
下载:
