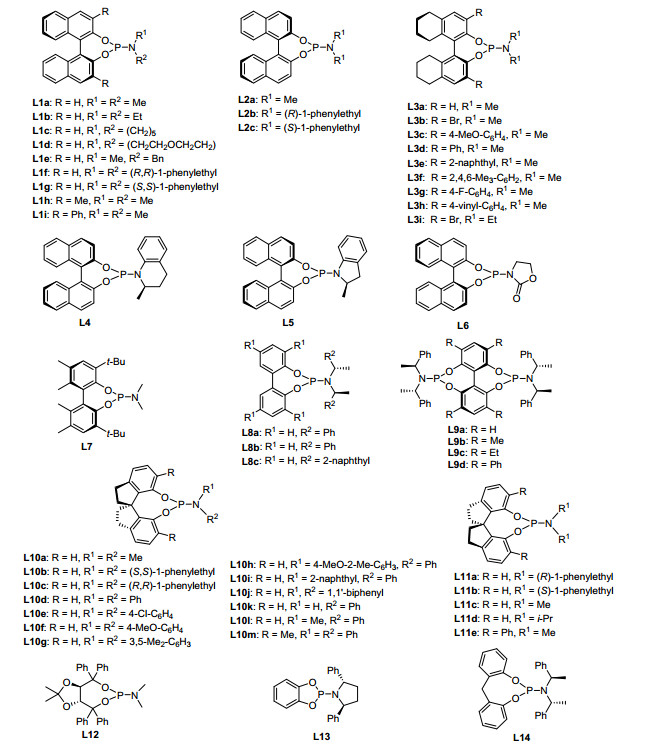
 图 1
手性亚磷酰胺配体举例
Figure 1.
Examples of chiral phosphoramidites
图 1
手性亚磷酰胺配体举例
Figure 1.
Examples of chiral phosphoramidites
引用本文:
袁乾家, 张万斌. 亚磷酰胺配体在铱催化不对称氢化反应中的应用[J]. 有机化学,
2015, 36(2): 274-282.
doi:
10.6023/cjoc201509016

Citation: Yuan Qianjia, Zhang Wanbin. Applications of Phosphoramidite Ligands in Ir-Catalyzed Asymmetric Hydrogenation Reactions[J]. Chinese Journal of Organic Chemistry, 2015, 36(2): 274-282. doi: 10.6023/cjoc201509016
Citation: Yuan Qianjia, Zhang Wanbin. Applications of Phosphoramidite Ligands in Ir-Catalyzed Asymmetric Hydrogenation Reactions[J]. Chinese Journal of Organic Chemistry, 2015, 36(2): 274-282. doi: 10.6023/cjoc201509016
亚磷酰胺配体在铱催化不对称氢化反应中的应用
English
Applications of Phosphoramidite Ligands in Ir-Catalyzed Asymmetric Hydrogenation Reactions
Abstract:
Phosphoramidites, as a class of privileged chiral ligands, are suitable for different types of reactions, such as catalytic asymmetric hydrogenation, catalytic asymmetric allylic substitution, catalytic asymmetric Diels-Alder reaction and so on. Catalytic asymmetric hydrogenation reactions are some of the most important reactions in industry. In this review recent advances and applications of phosphoramidite ligands in Ir-catalyzed asymmetric hydrogenation of enamides and their derivatives, unfunctionalized enamines, imines and heteroaromatic compounds are discussed.
-
Key words:
- phosphoramidites
- / iridium
- / catalytic asymmetric hydrogenation
-
过渡金属参与的不对称催化反应作为获得手性化合物的重要手段, 越来越受到人们的重视, 并成为化学工作者研究的热门领域之一[1~5].不对称催化反应的关键之一是手性配体的设计合成, 即配体很大程度上决定了不对称催化反应的效果.鉴于配体对不对称催化反应的重要性, 化学工作者经过几十年的努力已设计并合成了成千上万种配体.在如此繁多的手性配体中, 有一些配体可适用于多种类型的反应而被称为“优势手性配体”, 如具有联萘骨架的轴手性配体1, 1'-联萘-2, 2'-双二苯膦(BINAP)、1, 1'-联-2-萘酚(BINOL)和亚磷酰胺配体、面手性配体(R)-(-)-1-[(S)-2-二苯基磷]二茂铁基乙基二环己基磷(Josiphos)、中心手性配体(S, S)-(-)-2, 2'-亚异丙基双(4-叔丁基-2-噁唑啉(BOX)和(R)-(-)-2-[2-(二苯基膦)苯基]-4-苯基-2-噁唑啉(PHOX)、口袋型Salen配合物以及螺手性类配体等[6].
相比较于双配位和多配位型配体, 单配位型亚磷酰胺配体被开发利用较晚. 1994年Feringa课题组[7]首次报道了单配位型手性亚磷酰胺配体, 并在两年后将其成功地运用于铜催化1, 4-共轭加成反应中之后[8], 亚磷酰胺配体才引起了人们的重视.由于亚磷酰胺配体性质稳定、合成简便、易于修饰及效果独特等优点, 基于各种骨架类型的亚磷酰胺配体被先后报道, 如基于联萘二酚骨架的亚磷酰胺配体、基于联苯二酚骨架的亚磷酰胺配体、基于螺环骨架的亚磷酰胺配体、基于2, 2, 2', 2'-四芳基-1, 3-二氧戊环-4, 5-二甲醇骨架的亚磷酰胺配体及最近开发的D2-对称联苯基亚磷酰胺配体[9]等(图 1).亚磷酰胺配体在不对称催化共轭加成反应和不对称催化烯丙基取代反应方面的应用研究最近已经有综述文章[6b~6c]. 2006和2007年de Vries等[10]对亚磷酰胺配体在不对称催化氢化反应中的应用作了综述报道, 主要综述了单配位型亚磷酰胺配体在铑催化不对称氢化反应中的应用.鉴于单配位型亚磷酰胺配体在铱催化不对称氢化反应中的应用直到2007年才被首次报道[11], 该领域的文献综述至今还未见报道.另外, 基于铑的催化剂在对C=C双键进行不对称氢化时, 需要在底物C=C双键的邻近含有配位性官能团与催化剂中的金属铑配位以获得较高对映选择性的还原产物[1c].而基于铱的催化剂作用于C=C双键邻近不含配位性官能团底物时, 却往往可以获得很高对映选择性的氢化产物, 这使得基于铱的催化剂可以实现更广泛底物的不对称氢化[1f, 12].本文按照氢化底物类型(烯酰胺、烯胺、亚胺和芳香杂环)对单配位型亚磷酰胺配体在铱催化不对称氢化反应中的应用研究进行综述, 以利于人们对该领域进行全面的了解, 更好地推进该领域的应用发展.
图 1
手性亚磷酰胺配体举例
Figure 1.
Examples of chiral phosphoramidites
1 烯酰胺及其衍生物的不对称催化氢化
关于N-乙酰基烯胺的不对称催化氢化, 铑和钌双膦配合物以及铑/亚磷酰胺配合物已经取得了非常优异的成果[1], 铱/亚磷酰胺配合物对以下三类烯酰胺类底物的不对称催化氢化也取得了较为理想的结果: (1)烯酰胺类化合物; (2) α-脱氢氨基酸类化合物; (3) β-脱氢氨基酸类化合物, 如图 2所示.
 图 2
烯酰胺及其衍生物
Figure 2.
Enamides and their derivatives
图 2
烯酰胺及其衍生物
Figure 2.
Enamides and their derivatives
1.1 烯酰胺类化合物的不对称催化氢化
关于铱催化剂对简单烯酰胺的不对称氢化研究报道很少, 且反应的对映选择性较差(最高达60% ee)[13, 14].直到2009年, Beller等[15]将铱/亚磷酰胺配合物应用于该反应, 才取得了较为满意的结果(表 1).反应以基于H8-联萘二酚骨架的3, 3'-取代亚磷酰胺为配体, 详细地考察了配体3, 3'-位取代基团R对反应的影响(表 1, Entries 1~7).当3, 3'-位取代基团为H时, 基本上没有形成活性的催化剂(表 1, Entry 1);当R为4-甲氧基苯基时, 产物的对映选择性高达84%(表 1, Entry 3).当过渡金属铱与配体的物质的量的比从1:1增加到1:2时, 催化体系对反应的催化效果没有影响.他们也考察了不同阴离子的钠盐, 如HCOONa、Na2SO4、十二烷基磺酸钠(SDS)、NaBF4、NaClO4、NaPF6和NaBArF等对反应催化效果的影响.研究发现以NaClO4为最好, 可以获得93% ee的产物, 遗憾的是催化体系的适用性很差.当反应以单质碘作为添加剂时, 得到的氢化产物几乎是消旋的(2% ee).
 表 1
简单烯酰胺的不对称氢化
Table 1.
Asymmetric hydrogenation of simple enamides
表 1
简单烯酰胺的不对称氢化
Table 1.
Asymmetric hydrogenation of simple enamides

Entry L* Conv./% ee/% 1 L3a 14 — 2 L3b 27 18 3 L3c 100 84 4 L3d 85 56 5 L3e 100 78 6 L3f 60 44 7 L3g 100 82 表 1 简单烯酰胺的不对称氢化
Table 1. Asymmetric hydrogenation of simple enamides1.2 α-脱氢氨基酸衍生物的不对称催化氢化
2007年, de Vries等[11]报道了铱/亚磷酰胺配合物催化α-脱氢氨基酸衍生物2-乙酰氨基肉桂酸甲酯的不对称氢化, 这是首例一个单配位型配体与一个金属所形成的配合物在不对称催化氢化反应中的应用(表 2, Entries 1~4).增加配体联萘骨架3, 3'-位取代基团的空间位阻, 不但可以增加催化体系的反应活性, 而且所得产物的对映选择性也有明显提高.当3, 3'-位取代基团为苯基时, 氢化产物的对映选择性高达93%(表 2, Entry 3); 3, 3'-位为叔丁基取代的联苯亚磷酰胺配体L7应用于该反应, 产物的对映选择性可达98%, 并且催化体系的活性也有大幅提高(表 2, Entry 4).他们也考察了配体中氮原子上取代基团对反应的影响.发现当把配体L1a中二甲基胺基换为双(1-苯基乙基)胺基时, 催化体系的催化活性及对映选择性都很不理想.他们利用核磁和单晶衍射实验确认了过渡金属铱与亚磷酰胺配体的比例为1:1.
表 2
2-乙酰氨基肉桂酸甲酯的不对称氢化
Table 2.
Asymmetric hydrogenation of methyl 2-acetamido-cinnamate

Entry S/C a L* TOF/h-1 ee/% 1 50:1 L1a 6 28 2 50:1 L1h 24 67 3 50:1 L1i 50 93 4 50:1 L7 150 98 5 25:1 L3a — 37 6 25:1 L3b 11 91 7 25:1 L3c 17 94 8 25:1 L3d 25 96 9 25:1 L3e 22 95 10 25:1 L3f 24 98 11 25:1 L3g 14 95 12 25:1 L3h 20 94 13 25:1 L3i 16 89 14 50:1 L3f 37 98 15 b 50:1 L3f 37 99 a S/C=substrate/catalyst. b[Ir(cod)Cl]2/L*=1:4. 表 2 2-乙酰氨基肉桂酸甲酯的不对称氢化
Table 2. Asymmetric hydrogenation of methyl 2-acetamido-cinnamate2008年, Beller等[16]报道了基于H8-联萘骨架亚磷酰胺配体在铱催化α-脱氢氨基酸衍生物2-乙酰氨基肉桂酸甲酯不对称氢化反应中的应用(表 2, Entries 5~15).从表 2中可以得知, 在H8-联萘骨架3, 3'-位引入取代基团对反应的活性和对映选择性有着非常明显的影响.当3, 3'-位取代基团为大位阻2, 4, 6-三甲基苯基时, 氢化产物的对映选择性高达98%(表 2, Entry 10).配体L3d与L1i给出了几乎相当的不对称催化效果, 虽然这两种配体的骨架不同(表 2, Entry 8 vs Entry 3).当增加Ir/L3f=1:2时, 反应的催化效果与Ir/L3f=1:1时没有明显变化(表 2, Entry 15 vs Entry 14), 这间接表明了所形成的催化剂为单配位的铱配合物.
1.3 β-脱氢氨基酸衍生物的不对称催化氢化
2008年, Beller等[17]报道了基于H8-联萘骨架亚磷酰胺配体在铱催化β-脱氢氨基酸衍生物不对称氢化反应中的应用(表 3).与这类配体在铱催化α-脱氢氨基酸衍生物的不对称氢化(表 2)相似, 3, 3'-位取代基团对反应的催化效果起着决定性的作用.当3, 3'-位取代基团是4-位含有取代基团的苯基时, 氢化产物的对映选择性高达88%(表 3, Entries 3, 7和8).这类催化体系对(E)-β-脱氢氨基酸衍生物的不对称催化效果要明显优于对(Z)-β-脱氢氨基酸衍生物的不对称催化效果, 该类催化体系中也被证实Ir/L*=1:1.
表 3
β-脱氢氨基酸衍生物的不对称氢化
Table 3.
Asymmetric hydrogenation of β-dehydroamino acid derivatives

Entry L* Conv./% ee/% 1 L3a 53 60 2 L3b 55 84 3 L3c 100 88 4 L3d 71 84 5 L3e 100 86 6 L3f 99 74 7 L3g 99 88 8 L3h 100 88 9 L3i 99 84 表 3 β-脱氢氨基酸衍生物的不对称氢化
Table 3. Asymmetric hydrogenation of β-dehydroamino acid derivatives另外, 他们也考察了这类催化体系对四取代环内双键和环外双键β-脱氢氨基酸衍生物的不对称催化氢化效果.非常遗憾的是, 对这两类底物的不对称催化氢化只得到了痕量的还原产物(Scheme 1).
 图 图式1
四取代β-脱氢氨基酸衍生物的不对称氢化
Figure 图式1.
Asymmetric hydrogenation of tetrasubstituted β-dehydroamino acid derivatives
图 图式1
四取代β-脱氢氨基酸衍生物的不对称氢化
Figure 图式1.
Asymmetric hydrogenation of tetrasubstituted β-dehydroamino acid derivatives
2 非官能团烯胺的不对称催化氢化
不同于N-乙酰基烯胺类化合物, 非官能团烯胺类化合物由于C=C双键附近没有配位性的官能团与催化剂的金属中心进行配位, 所以这类底物的不对称催化氢化很难由基于过渡金属铑和钌的催化剂来实现.这类底物的高效不对称氢化的例子也因之报道较少[18~22].基于过渡金属铱的催化剂对C=C双键进行不对称催化氢化时, 在底物C=C双键附近没有配位性官能团, 也能很好地控制产物的对映选择性, 这使得基于铱的催化剂可以实现更广泛烯烃的不对称催化氢化[12a].
2009年, 周其林等[20]在单质碘为添加剂、铱:亚磷酰胺=1:2的催化体系下实现了五元环状烯胺的不对称氢化(表 4).从表 4中可以知道, 亚磷酰胺配体中氮原子上取代基团的空间位阻以及骨架手性和氮原子上取代基团中心手性的匹配对催化产物的对映选择性有着非常明显的影响(表 4, Eentries 1~5).研究表明, (Ra, S, S)构型螺手性配体L10b为反应的最优配体(表 4, Entry 4).氢气压力对产物的对映选择性也有一定的影响, 当氢气压力从5.06 MPa降至101 kPa时, 产物的对映选择性由92%升至94%.双膦配体如: (R)-(+)-7, 7'-双(二苯基磷)-2, 2', 3, 3'-四氢-1, 1'-螺环茚烷(SDP)、(S)-(-)-1, 1'-联萘-2, 2'-双二苯膦(BINAP)、(R)-(-)-1-[(S)-2-二苯基磷]二茂铁基乙基二环己基磷(Josiphos)、(-)-1, 2-双((2R, 5R)-2, 5-二甲基磷)苯(Me-Duphos)和R-(+)-6, 6'-双(二苯基磷)-2, 2', 3, 3'-四氢-5, 5'-二-1, 4-苯并二辛烷(Synphos)对这类底物的不对称催化氢化未能表现出令人满意的结果.他们将所开发的催化体系成功的运用于异喹啉类生物碱Crispine A的不对称合成中(Eq. 1).这类催化体系只适用于五元环状烯胺, 对六元环状烯胺的不对称氢化所得产物的对映选择性最高只有20%.
表 4
环状烯胺的不对称氢化
Table 4.
Asymmetric hydrogenation of cyclic enamines

Entry L* Conv./% ee/% (Conf.) 1 L10a 100 44 (S) 2 L1f 100 88 (R) 3 L1g 100 70 (S) 4 L10b 100 92 (S) 5 L10c 100 57 (S) 表 4 环状烯胺的不对称氢化
Table 4. Asymmetric hydrogenation of cyclic enamines2009年, 周其林等[21]报道了通过不对称催化氢化环外双键烯胺类底物合成四氢异喹啉类化合物的方法(表 5).当亚磷酰胺配体胺基部分是手性双(1-苯基乙基)胺基时, 基于螺环骨架和联萘骨架的亚磷酰胺配体均能给出很好的催化活性(表 5, Entries 1, 2, 6和7), 其中手性匹配的配体给出较好的对映选择性(表 5, Entry 1 vs Entry 2, Entry 6 vs Entry 7).当亚磷酰胺配体胺基氮原子上的取代基团为简单的烷基时, 催化体系的反应活性和对映选择性都有非常明显的降低(表 5, Etries 3~5).他们对配体与金属的配位情况进行了考察, 当Ir: L11a=1:1时, 反应的速率较低, 氢化产物的对映选择性为87%;当Ir:L11a=1:2时, 反应的速率明显提高而且氢化产物的对映选择性提高至95%;继续增大Ir:L11a=1:3对反应的速率和氢化产物的对映选择性没有影响.基于添加剂碘化钾和单质碘对此类反应有相同的活化作用, 结合相关的实验数据他们提出了反应经过了Ir-双氢物种的Ir(Ⅰ)-Ir(Ⅲ)催化循环(Scheme 2).首先, 在过量单质碘或碘化钾存在下, 一个金属铱与两分子亚磷酰胺配体形成Ir(Ⅰ)-配合物B或其二聚体C. Ir(Ⅰ)-配合物B与氢气经过氧化加成形成Ir(Ⅲ)-二氢物种D, Ir(Ⅲ)-二氢物种D中的金属原子与底物烯胺中的C=C双键以Ƞ2的形式配位形成复合物E.然后金属原子上的一个氢转移至烯胺底物中与N原子相邻的不饱和碳原子上形成复合物F.最后复合物F经过还原消除得到氢化产物并给出Ir(Ⅰ)-配合物B, 完成催化循环.另外, 基于铱的催化体系对亚胺和喹啉的不对称氢化时, 单质碘或碘化物对反应的催化活性和对映选择性有着非常重要的影响, 他们提出了包含Ir(Ⅲ)-单氢物种的催化循环过程[23].
表 5
四氢异喹啉类化合物的合成
Table 5.
Synthesis of tetrahydroisoquinolines

Entry L* Conv./% ee/% (Conf.) 1 L11a 100 95 (R) 2 L11b 100 62 (R) 3 L11c 17 40 (R) 4 L11d 78 54 (R) 5 L11e 3 25 (R) 6 L1f 100 94 (R) 7 L1g 100 80 (S) 表 5 四氢异喹啉类化合物的合成
Table 5. Synthesis of tetrahydroisoquinolines 图 图式2
可能的催化循环
Figure 图式2.
Proposed catalytic cycle
图 图式2
可能的催化循环
Figure 图式2.
Proposed catalytic cycle
2010年, 周其林等[22]报道了铱/亚磷酰胺配合物催化N, N-二烷基简单烯胺的不对称氢化(表 6).考察了亚磷酰胺配体胺基氮原子上不同芳基取代的配体(表 6, Entries 1~9), 获得高达87%的对映选择性(表 6, Entries 1~2).而在配体的螺环骨架上引入甲基, 所得氢化产物的对映选择性只有27%(表 6, Entry 10).此催化体系只对胺基为五元环状胺基类底物有较好的不对称催化效果, 另外, 他们也考察了催化体系对于环状N, N-二烷基烯胺类底物的不对称氢化(表 7).对于不同大小环状的底物取得了80~90% ee(表 7, Entries 1~3), 七元环底物获得了最高90%的对映选择性(表 7, Entry 3).
表 6
N, N-二烷基烯胺的不对称氢化
Table 6.
Asymmetric hydrogenation of N, N-dialkylenamines

Entry L* Conv./% ee/% 1 L10d 100 87 2 L10e 100 87 3 L10f 100 78 4 L10g 36 17 5 L10h 100 75 6 L10i 100 85 7 L10j 100 70 8 L10k 67 7 9 L10l 30 15 10 L10m 100 27 表 6 N, N-二烷基烯胺的不对称氢化
Table 6. Asymmetric hydrogenation of N, N-dialkylenamines
表 7
环内双键烯胺的不对称氢化
Table 7.
Asymmetric hydrogenation of enamines with endo-double bond

Entry n Conv./% ee/% 1 1 100 80 2 2 100 88 3 3 100 90 表 7 环内双键烯胺的不对称氢化
Table 7. Asymmetric hydrogenation of enamines with endo-double bond3 亚胺的不对称催化氢化
基于过渡金属铑、铱、钌和钛的催化剂对亚胺的不对称氢化都取得了很好的效果[1], 其中基于铱的双膦配体催化体系和P, N-配体催化体系是最为突出的, 例如Ir/Xyliphos催化体系已经应用于除草剂异丙甲草胺的工业化生产[24].目前只有三例铱/亚磷酰胺催化体系在亚胺的不对称氢化反应中的应用, 分别是对N-芳基亚胺、N-H亚胺和环状亚胺的不对称氢化[25~27].
3.1 N-芳基亚胺的不对称催化氢化
2009年, de Vries等[25]实现了单配位型亚磷酰胺配体在亚胺不对称催化氢化中的应用, 其中铱:亚磷酰胺配体=1:2(表 8).铱盐中的阴离子对催化体系的活性有非常明显的影响, 当阴离子为BArF-和PF6-时, 反应可以完全进行并给出中等的对映选择性(表 8, Entries 1~2);而当阴离子为Cl-时, 则得不到氢化产物(表 8, Entry 3).升高反应温度至60 ℃、氢气压力至5.06 MPa, 该反应才可以完全进行(表 8, Entry 4).当反应以[Ir(cod)2]BArF为铱盐, 氢气压力为101 kPa时, 反应完全进行并得到87% ee的氢化产物.
表 8
N-芳基亚胺的不对称氢化
Table 8.
Asymmetric hydrogenation of N-arylimines

Entry Ir salt Conv./% ee/% 1 [Ir(cod)2]BArF 100 80 2 [Ir(cod)2]PF6 100 65 3 [Ir(cod)Cl]2 0 — 4 a [Ir(cod)Cl]2 100 61 a Reaction performed at 60 ℃, 5.06 MPa H2. 表 8 N-芳基亚胺的不对称氢化
Table 8. Asymmetric hydrogenation of N-arylimines3.2 N-H亚胺的不对称催化氢化
2009年, Zhang和Gosselin等[26]报道了铱/亚磷酰胺催化取代二苯甲酮N-H亚胺不对称氢化反应(表 9).由于铱/双膦催化体系在N-H亚胺不对称氢化反应中表现出了很好的效果[28], 他们首先筛选了双膦配体(f-Binaphane, Segphos, BINAP和Josiphos)在铱催化取代二苯甲酮N-H亚胺不对称氢化中的应用, 但均没有得到理想的结果(up to 47% conv., 64% ee).他们对单配位型亚磷酰胺配体进行了筛选(表 9, Entries 1~10), 发现以L1e为配体, 10.34 Mpa H2氛围下, 反应可以完全进行并且所得氢化产物的对映选择性高达83%(表 9, Entry 9);当MeOH:DCM=1:3时, 反应可以获得87% ee的氢化产物(表 9, Entry 10).底物芳基邻位取代基团对反应所得产物的对映选择性起着至关重要的作用, 当芳基邻位取代基团为CF3-时, 氢化产物的对映选择性高达98%.
表 9
取代二苯甲酮N-H亚胺的不对称氢化
Table 9.
Asymmetric hydrogenation of substituted benzophenone N-H imines

Entry L* Conv./% ee/% 1 L11c 100 62 2 L10c 20 20 3 L2a 87 66 4 L1b 70 80 5 L1d 85 76 6 L1c 62 83 7 L1f 100 72 8 L1g 100 80 9 a L1e 100 83 10 a , b L1e 100 87 a Reaction performed at 10.34 MPa. b V(MeOH):V(DCM)=1:3. 表 9 取代二苯甲酮N-H亚胺的不对称氢化
Table 9. Asymmetric hydrogenation of substituted benzophenone N-H imines3.3 环状亚胺的不对称催化氢化
2012年, 周其林等[27]实现了通过铱/亚磷酰胺催化环状亚胺不对称氢化来合成四氢异喹啉类化合物(表 10).从表 10中可以知道, 对于这类底物的不对称催化氢化, 基于螺环骨架亚磷酰胺配体催化效果明显优于基于联苯二酚骨架亚磷酰胺配体(表 10, Entry 1 vs Entries 12和3).基于联苯二酚骨架亚磷酰胺配体中, 轴手性与胺基部分中心手性的匹配性对反应的催化活性有着非常明显的影响(表 10, Entry 2 vs Entry 3).利用作者所开发的催化体系可以方便的进行四环类生物碱番荔枝宁[(S)-xylopinine]的不对称合成(Scheme 3).
表 10
环状亚胺的不对称氢化
Table 10.
Asymmetric hydrogenation of cyclic imines

Entry L* Conv./% ee/% (Conf.) 1 L10b 100 99 (S) 2 L1f 22 82 (R) 3 L1g 95 87 (S) 表 10 环状亚胺的不对称氢化
Table 10. Asymmetric hydrogenation of cyclic imines 图 图式3
番荔枝宁的合成
Figure 图式3.
Synthesis of (S)-xylopinine
图 图式3
番荔枝宁的合成
Figure 图式3.
Synthesis of (S)-xylopinine
4 芳香杂环化合物的不对称催化氢化
关于含氮芳香杂环2-取代喹啉的不对称催化氢化已有很多催化体系被报道[29].例如铱/双膦配体催化体系[30]、铱/P, N-配体催化体系[31]、铱/双氧磷配体催化体系[32]和铱/双亚磷酸酯/非手性单膦催化体系[33]都在反应中取得了很好的效果.另外, 基于铱/双膦催化体系的不对称转移氢化[34]和手性磷酸催化的不对称转移氢化[35]也都取得了非常好的结果.
2008年, de Vries等[36]报道了铱/亚磷酰胺配体/非手性单膦配体催化体系对2-取代喹啉不对称氢化反应的研究, 此催化体系必须以哌啶盐酸盐作为添加剂.非手性单膦配体对反应的活性和产物的对映选择性有非常明显的影响(表 11).以三苯基膦为反应中非手性单膦配体时, 反应几乎没有进行(表 11, Entry 1);邻位有取代基团的三芳基膦配体和三叔丁基膦配体可以给出较好的转化率和对映选择性(表 11, Entries 2~7), 其中以三(邻甲基苯基)膦为最好(表 11, Entry 2).
表 11
喹喔啉的不对称氢化
Table 11.
Asymmetric hydrogenation of quinolines

Entry Phosphine Conv./% ee/% 1 PPh3 2 — 2 P(2-MeC6H4)3 100 83 3 P(2, 4, 6-Me3C6H2)3 100 70 4 P(2-Naphthyl)3 41 81 5 P(t-Bu)3 54 82 6 P(2-MeOC6H4)3 18 77 7 P(C6F5)3 98 78 表 11 喹喔啉的不对称氢化
Table 11. Asymmetric hydrogenation of quinolines在铱/L1c/P(2-Me-C6H4)3催化体系对喹啉的不对称氢化基础上, 2009年de Vries等[37]报道了铱/L1c催化喹喔啉的不对称氢化, 得到的氢化产物最高达96% ee (Eq. 2).
5 结论与展望
本文主要综述了单配位型亚磷酰胺配体在铱催化不对称氢化方面的应用研究.经过短短几年的发展, 单配位型亚磷酰胺配体在铱催化不对称氢化反应中的应用已经取得了一定的效果.尤其是一个金属与一个单配位型亚磷酰胺配体所形成的催化体系在不对称氢化中的成功应用, 打破了人们几十年来对只有双配位型配体才能很好地控制过渡金属催化不对称氢化反应的局限.另外, 铱/亚磷酰胺催化体系对一些底物的不对称氢化表现出了绝对的优势.目前, 被报道的基于铱/亚磷酰胺催化体系的不对称氢化反应都集中于含氮原子的底物, 对其他类型底物(如羰基、简单烯烃等)的不对称氢化有待进一步开发.除了本文所综述的单配位型亚磷酰胺配体在铱催化不对称氢化方面的应用之外, 基于亚磷酰胺所开发的磷-亚磷酰胺配体[38]、氮-亚磷酰胺配体[39]在铱催化不对称氢化反应中均表现出了很好的效果.由于亚磷酰胺配体性质稳定、易于合成与修饰和效果独特等优点, 我们相信将会有更多更好的关于亚磷酰胺配体在铱催化不对称氢化反应中的研究成果被报道.
-
-
[1]
Noyori, R. Angew. Chem., Int. Ed. 2002, 41, 2008.
(b) Lu, S.-M.; Han, X.-W.; Zhou, Y.-G. Chin. J. Org. Chem. 2005, 25, 634 (in Chinese).
(卢胜梅, 韩秀文, 周永贵, 有机化学, 2005, 25, 634.)
(c) de Vries, J. G.; Elsevier, C. J. Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2007.
(d) Ma, Y.-H.; Zhang, Y.-J.; Zhang, W. Chin. J. Org. Chem. 2007, 27, 289 (in Chinese).
(马元辉, 张勇健, 张万斌, 有机化学, 2007, 27, 289.)
(e) Blaser, H. U.; Federsel. H.-J. Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, 2nd ed., Wiley-VCH, Weinheim, 2010.
(f) Xie, J.-H.; Zhu, S.-F.; Zhou, Q.-L. Chem. Rev. 2011, 111, 1713. -
[2]
Trost, B. M.; Crawley, M. L. Chem. Rev. 2003, 103, 2921.
(b) Lu, Z.; Ma, S. Angew. Chem., Int. Ed. 2008, 47, 258.
(c) Zhang, W.; Liu, D. In Chiral Ferrocenes in Asymmetric Catalysis: Synthesis and Applications, Chapter 7, Wiley-VCH, Weinheim, 2010.
(d) Kazmaier, U. Transition Meatl Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, Springer: New York, 2012.
(e) Zhuo, C.-X.; Zheng, C.; You, S.-L. Acc. Chem. Res. 2014, 47, 2558. -
[3]
Kagan, H. B.; Riant, O. Chem. Rev. 1992, 92, 1007.
(b) Kobayashi, S.; Jorgenson, K. A. Cycloaddition Reaction in Organic Synthesis, New York, Wiley-VCH, 2002.
(c) Desimoni, G.; Faita, G.; Jorgenson, K. A. Chem. Rev. 2006, 106, 3561. -
[4]
Porter, M. J.; Skidmore, J. Chem. Commun. 2000, 36, 1215.
(b) Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Chem. Rev. 2005, 105, 1603. -
[5]
RajanBabu, T. V. Chem. Rev. 2003, 103, 2845.
(b) Jia, X.; Wang, Z.; Xia, C.; Ding, K. C. Chin. J. Org. Chem. 2013, 33, 1369 (in Chinese) (贾肖飞, 王正, 夏春谷, 丁奎岭, 有机化学, 2013, 33, 1369.) -
[6]
Yoon, T. P.; Jacobsen, E. N. Science 2003, 299, 1691.
(b) Teichert, J. F.; Feringa, B. L. Angew. Chem., Int. Ed. 2010, 49, 2486.
(c) Zhang, Z.; Xie, F.; Yang, B.; Yu, H.; Zhang, W. Chin. J. Org. Chem. 2011, 31, 429 (in Chinese).
(张振锋, 谢芳, 杨波, 余焓, 张万斌, 有机化学, 2011, 31, 429.)
(d) Zhou, Q.-L. Privileged Chiral Ligands and Catalysts, Wiley-VCH: Weinheim, 2011. -
[7]
Hulst, R.; de Vries, N. K.; Feringa, B. L. Tetrahedron: Asymmetry 1994, 5, 699. doi: 10.1016/0957-4166(94)80032-4
-
[8]
de Veris, A. H. M.; Meetsma, A.; Feringa, B. L. Angew. Chem. Int. Ed. Engl. 1996, 35, 2374. doi: 10.1002/(ISSN)1521-3773
-
[9]
Zhang, H.; Fang, F.; Xie, F.; Yu, H.; Yang, G.; Zhang, W. Tetrahedron Lett. 2010, 51, 3119.
(b) Yang, B.; Xie, F.; Yu, H.; Shen, K.; Ma, Z.; Zhang, W. Tetrahedron 2011, 67, 6197.
(c) Yu, H.; Xie, F.; Ma, Z.; Liu, Y.; Zhang, W. Org. Biomol. Chem. 2012, 10, 5137.
(d) Yu, H.; Xie, F.; Ma, Z.; Liu, Y.; Zhang, W. Adv. Synth. Catal. 2012, 354, 1941. -
[10]
Minnaard, A. J.; Feringa, B. L.; Lefort, L.; de Vries, J. G. Acc. Chem. Res. 2007, 40, 1267.
(b) de Vries, A. H. M.; de Vries, J. G. Platinum Metals Rev. 2006, 50, 54. -
[11]
Giacomina, F.; Meetsma, A.; Panella, L.; Lefort, L.; de Vries, A. H. M.; de Vries, J. G. Angew. Chem., Int. Ed. 2007, 46, 1497. doi: 10.1002/(ISSN)1521-3773
-
[12]
Roseblade, S. J.; Pfaltz, A. Acc. Chem. Res. 2007, 40, 1402.
(b) Zhang, J.; Yang, D.; Long, Y. Chin. J. Org. Chem. 2009, 29, 835 (in Chinese).
(张俊芳, 杨定乔, 龙玉华, 有机化学, 2009, 29, 835.) -
[13]
Marie, P.; Deblon, S.; Breher, F.; Geier, J.; Böhler, C.; Rüegger, H.; Schönberg, H.; Grützmacher, H. Chem. Eur. J. 2004, 10, 4198. doi: 10.1002/(ISSN)1521-3765
-
[14]
Broady, S. D.; Martin, D. M. G.; Lennon, I. C.; Ramsden, J. A.; Muir, J. C. WO 2006067412, 2006 [Chem. Abstr. 2006, 145, 103874].
-
[15]
Erre, G.; Enthaler, S.; Junge, K.; Addis, D.; Beller, M. Adv. Synth. Catal. 2009, 351, 1437. doi: 10.1002/adsc.v351:9
-
[16]
Erre, G.; Junge, K.; Enthaler, S; Addis, D.; Michalik, D.; Spannenberg, A.; Beller, M. Chem. Asian J. 2008, 3, 887. doi: 10.1002/(ISSN)1861-471X
-
[17]
Enthaler, S.; Erre, G.; Junge, K.; Schröder, K.; Addis, D.; Michalik, D.; Hapke, M.; Redkin, D.; Beller, M. Eur. J. Org. Chem. 2008, 3352.
-
[18]
Lee, N. E.; Buchwald, S. L. J. Am. Chem. Soc. 1994, 116, 5985. doi: 10.1021/ja00092a066
-
[19]
Hou, G.-H.; Xie, J.-H.; Wang, L.-X.; Zhou, Q.-L. J. Am. Chem. Soc. 2006, 128, 11774. doi: 10.1021/ja0644778
-
[20]
Hou, G.-H.; Xie, J.-H.; Yan, P.-C.; Zhou, Q.-L. J. Am. Chem. Soc. 2009, 131, 1366. doi: 10.1021/ja808358r
-
[21]
Yan, P.-C.; Xie, J.-H.; Hou, G.-H.; Wang, L.-X.; Zhou, Q.-L. Adv. Synth. Catal. 2009, 351, 3243. doi: 10.1002/adsc.v351:18
-
[22]
Yan, P.-C.; Xie, J.-H.; Zhou, Q.-L. Chin. J. Chem. 2010, 28, 1736. doi: 10.1002/cjoc.201090293
-
[23]
Xiao, D.; Zhang, X. Angew. Chem., Int. Ed. 2001, 40, 3425.
(b) Wang, D.-W.; Wang, X.-B.; Wang, D.-S.; Lu, S.-M.; Zhou, Y.-G.; Li, Y.-X. J. Org. Chem. 2009, 74, 2780. -
[24]
Blaser, H. U.; Buser, H. P.; Losers, K.; Hanreich, R.; Jalett, H. P.; Jelsch, E.; Pugin, B.; Schneider, H. D.; Splinder, F.; Wagmann, A. Chimia 1999, 53, 275.
-
[25]
Mršić, N.; Minnaard, A. J.; Feringa, B. L.; de Vries, J. G. J. Am. Chem. Soc. 2009, 131, 8358. doi: 10.1021/ja901961y
-
[26]
Hou, G.; Tao, R.; Sun, Y.; Zhang, X.; Gosselin, F. J. Am. Chem. Soc. 2010, 132, 2124. doi: 10.1021/ja909583s
-
[27]
Xie, J.-H.; Yan, P.-C.; Zhang, Q.-Q.; Yuan, K.-X.; Zhou, Q.-L. ACS Catal. 2012, 2, 561. doi: 10.1021/cs300069g
-
[28]
Hou, G.; Gosselin, F.; Li, W.; McWilliams, C.; Sun, Y.; Weisel, M.; O'Shea, P. D.; Chen, C.-Y.; Davies, I. W.; Zhang, X. J. Am. Chem. Soc. 2009, 131, 9882. doi: 10.1021/ja903319r
-
[29]
Wang, D.-S.; Chen, Q.-A.; Lu, S.-M.; Zhou, Y.-G. Chem. Rev. 2011, 112, 2557.
-
[30]
Wang, W.-B.; Lu, S.-M.; Yang, P.-Y.; Han, X.-W.; Zhou, Y.-G. J. Am. Chem. Soc. 2003, 125, 10536. doi: 10.1021/ja0353762
-
[31]
Lu, S.-M.; Han, X.-W.; Zhou, Y.-G. Adv. Synth. Catal. 2004, 346, 909. doi: 10.1002/(ISSN)1615-4169
-
[32]
Lam, K. H.; Lu, L.; Feng, L.; Fan, Q.-H.; Lam, F. L.; Lo, W.-H.; Chan, A. S. C. Adv. Synth. Catal. 2005, 347, 1755. doi: 10.1002/(ISSN)1615-4169
-
[33]
Reetz, M. T.; Li, X. Chem. Commun. 2006, 42, 2159.
-
[34]
Wang, D.-W.; Zeng, W; Zhou, Y.-G. Tetrahedron: Asymmetry 2007, 18, 1103. doi: 10.1016/j.tetasy.2007.04.028
-
[35]
Rueping, M.; Antonchick, A. P.; Theissmann, T. Angew. Chem., Int. Ed. 2006, 45, 3683. doi: 10.1002/(ISSN)1521-3773
-
[36]
Mršić, N.; Lefort, L.; Boogers, J. A. F.; Minnaard, A. J.; Feringa, B. L.; de Vries, J. G. Adv. Synth. Catal. 2008, 350, 1081. doi: 10.1002/(ISSN)1615-4169
-
[37]
Mršić, N.; Jerphagnon, T.; Minnaard, A. J.; Feringa, B. L.; de Vries, J. G. Adv. Synth. Catal. 2009, 351, 2549. doi: 10.1002/adsc.200900522
-
[38]
Eggenstein, M.; Thomas, A.; Theuerkauf, J.; Franciò, G.; Leitner, W. Adv. Synth. Catal. 2009, 351, 725.
(b) Hou, C.-J.; Wang, Y.-H.; Zheng, Z.; Xu, J.; Hu, X.-P. Org. Lett. 2012, 14, 3554. -
[39]
Bess, E. N.; Sigman, M. S. Org. Lett. 2013, 15, 646.
(b) Biosca, M.; Paptchikhine, A.; Pàmies, O.; Andersson, P. G.; Diéguez, M. Chem. Eur. J. 2015, 21, 3455.
-
[1]
-

图式1 四取代β-脱氢氨基酸衍生物的不对称氢化
Scheme 1 Asymmetric hydrogenation of tetrasubstituted β-dehydroamino acid derivatives
表 1 简单烯酰胺的不对称氢化
Table 1. Asymmetric hydrogenation of simple enamides
Entry L* Conv./% ee/% 1 L3a 14 — 2 L3b 27 18 3 L3c 100 84 4 L3d 85 56 5 L3e 100 78 6 L3f 60 44 7 L3g 100 82  下载: 导出CSV
下载: 导出CSV
表 2 2-乙酰氨基肉桂酸甲酯的不对称氢化
Table 2. Asymmetric hydrogenation of methyl 2-acetamido-cinnamate
Entry S/C a L* TOF/h-1 ee/% 1 50:1 L1a 6 28 2 50:1 L1h 24 67 3 50:1 L1i 50 93 4 50:1 L7 150 98 5 25:1 L3a — 37 6 25:1 L3b 11 91 7 25:1 L3c 17 94 8 25:1 L3d 25 96 9 25:1 L3e 22 95 10 25:1 L3f 24 98 11 25:1 L3g 14 95 12 25:1 L3h 20 94 13 25:1 L3i 16 89 14 50:1 L3f 37 98 15 b 50:1 L3f 37 99 a S/C=substrate/catalyst. b[Ir(cod)Cl]2/L*=1:4.
下载: 导出CSV
表 3 β-脱氢氨基酸衍生物的不对称氢化
Table 3. Asymmetric hydrogenation of β-dehydroamino acid derivatives
Entry L* Conv./% ee/% 1 L3a 53 60 2 L3b 55 84 3 L3c 100 88 4 L3d 71 84 5 L3e 100 86 6 L3f 99 74 7 L3g 99 88 8 L3h 100 88 9 L3i 99 84
下载: 导出CSV
表 4 环状烯胺的不对称氢化
Table 4. Asymmetric hydrogenation of cyclic enamines
Entry L* Conv./% ee/% (Conf.) 1 L10a 100 44 (S) 2 L1f 100 88 (R) 3 L1g 100 70 (S) 4 L10b 100 92 (S) 5 L10c 100 57 (S)
下载: 导出CSV
表 5 四氢异喹啉类化合物的合成
Table 5. Synthesis of tetrahydroisoquinolines
Entry L* Conv./% ee/% (Conf.) 1 L11a 100 95 (R) 2 L11b 100 62 (R) 3 L11c 17 40 (R) 4 L11d 78 54 (R) 5 L11e 3 25 (R) 6 L1f 100 94 (R) 7 L1g 100 80 (S)
下载: 导出CSV
表 6 N, N-二烷基烯胺的不对称氢化
Table 6. Asymmetric hydrogenation of N, N-dialkylenamines
Entry L* Conv./% ee/% 1 L10d 100 87 2 L10e 100 87 3 L10f 100 78 4 L10g 36 17 5 L10h 100 75 6 L10i 100 85 7 L10j 100 70 8 L10k 67 7 9 L10l 30 15 10 L10m 100 27
下载: 导出CSV
表 7 环内双键烯胺的不对称氢化
Table 7. Asymmetric hydrogenation of enamines with endo-double bond
Entry n Conv./% ee/% 1 1 100 80 2 2 100 88 3 3 100 90
下载: 导出CSV
表 8 N-芳基亚胺的不对称氢化
Table 8. Asymmetric hydrogenation of N-arylimines
Entry Ir salt Conv./% ee/% 1 [Ir(cod)2]BArF 100 80 2 [Ir(cod)2]PF6 100 65 3 [Ir(cod)Cl]2 0 — 4 a [Ir(cod)Cl]2 100 61 a Reaction performed at 60 ℃, 5.06 MPa H2.
下载: 导出CSV
表 9 取代二苯甲酮N-H亚胺的不对称氢化
Table 9. Asymmetric hydrogenation of substituted benzophenone N-H imines
Entry L* Conv./% ee/% 1 L11c 100 62 2 L10c 20 20 3 L2a 87 66 4 L1b 70 80 5 L1d 85 76 6 L1c 62 83 7 L1f 100 72 8 L1g 100 80 9 a L1e 100 83 10 a , b L1e 100 87 a Reaction performed at 10.34 MPa. b V(MeOH):V(DCM)=1:3.
下载: 导出CSV
表 10 环状亚胺的不对称氢化
Table 10. Asymmetric hydrogenation of cyclic imines
Entry L* Conv./% ee/% (Conf.) 1 L10b 100 99 (S) 2 L1f 22 82 (R) 3 L1g 95 87 (S)
下载: 导出CSV
表 11 喹喔啉的不对称氢化
Table 11. Asymmetric hydrogenation of quinolines
Entry Phosphine Conv./% ee/% 1 PPh3 2 — 2 P(2-MeC6H4)3 100 83 3 P(2, 4, 6-Me3C6H2)3 100 70 4 P(2-Naphthyl)3 41 81 5 P(t-Bu)3 54 82 6 P(2-MeOC6H4)3 18 77 7 P(C6F5)3 98 78
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 87
- HTML全文浏览量: 12
 下载:
下载:
