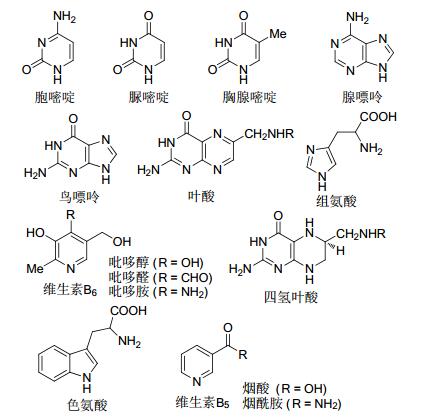
 图1
生命体中的含氮杂环化合物
Figure1.
Selected examples on N-heterocyclic compounds in living organisms
图1
生命体中的含氮杂环化合物
Figure1.
Selected examples on N-heterocyclic compounds in living organisms
引用本文:
廖云, 朱磊, 俞颖华, 陈贵, 黄学良. 金催化分子间炔烃的氮烯转移反应合成含氮杂环化合物[J]. 有机化学,
2017, 37(11): 2785-2799.
doi:
10.6023/cjoc201708021

Citation: Liao Yun, Zhu Lei, Yu Yinghua, Chen Gui, Huang Xueliang. N-Heterocycle Synthesis via Gold-Catalyzed Intermolecular Nitrene Transfer Reactions of Alkynes[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2785-2799. doi: 10.6023/cjoc201708021
Citation: Liao Yun, Zhu Lei, Yu Yinghua, Chen Gui, Huang Xueliang. N-Heterocycle Synthesis via Gold-Catalyzed Intermolecular Nitrene Transfer Reactions of Alkynes[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2785-2799. doi: 10.6023/cjoc201708021
金催化分子间炔烃的氮烯转移反应合成含氮杂环化合物
English
N-Heterocycle Synthesis via Gold-Catalyzed Intermolecular Nitrene Transfer Reactions of Alkynes
Abstract:
N-Heterocyclic rings are versatile structural units that widely dispersed in a variety of natural products, biological active species, and photoelectric materials. They are also useful building blocks in synthetic community. This review mainly focused on recent progress on gold-catalyzed intermolecular nitrene transfer reactions with alkynes. This strategy complements the toolbox for the synthesis of multisubstituted N-heterocyclic compounds. Mechanistically, activated by a suitable gold catalyst, the specific alkyne could react with a nitrene precursor, providing the final N-heterocyclic compounds in highly efficient manner.
-
Key words:
- gold-catalysis
- / ynamide
- / nitrene precursor
- / N-heterocycle
- / α-imino-gold-carbene
-
含氮杂环化合物无论是在治疗人类疾病的药物及杀灭虫害杂草的农药等具有生物和医药活性的化合物中, 还是在自然界丰富的天然产物中, 亦或是在一些液晶材料、荧光材料、染料等功能材料中都占据着非常大的比例, 也都扮演着非常重要的角色[1].比如生物体活细胞中DNA和RNA的基本组成单元就是胞嘧啶、尿嘧啶、胸腺嘧啶和鸟嘌呤、腺嘌呤等这些含氮杂环化合物; 氨基酸是生命体中蛋白质的基本组成单元, 而部分氨基酸也含有氮杂环结构片段, 如色氨酸、组氨酸; 对人体非常重要的一些维生素如维生素B5、维生素B6, 防止胎儿畸形的叶酸等, 也都是由一些特殊的氮杂环化合物构建而成的; 麻醉剂吗啡, 抗肿瘤药物长春花碱及长春花新碱等衍生物, 头孢类抗生素药物, 蛋白质、酶、癌细胞等的抑制剂等, 都具有含氮杂环的结构单元(图 1).所以, 长期以来发展高效构建含氮杂环的新方法都是有机合成领域的研究热点之一.
图1
生命体中的含氮杂环化合物
Figure1.
Selected examples on N-heterocyclic compounds in living organisms
自从近代化学发展以来, 含氮杂环化合物的合成方法得到了长足的发展.耳熟能详的人名反应不胜枚举, 如: Fischer吲哚合成、Bischler-Napieralski异喹啉合成、Paal-Knorr吡咯合成、Kröhnke吡啶合成等为含氮杂环的合成提供了多种快捷的合成路线.过渡金属催化合成含氮杂环化合物也是一种非常强有力的合成手段.近年来, 均相金催化发展迅速[2], 通过金催化炔烃的氮烯转移反应合成含氮杂环化合物也取得了突破性进展[3]. Toste, Zhang, Gagosz, Yamamoto等小组[4]分别使用金催化分子内炔烃的氮烯转移反应策略合成了吡咯、吲哚、喹啉、异喹啉、咪唑、吡啶等含氮杂环化合物及其衍生物(Scheme 1).但是在2010年以前, 有关金催化分子间炔烃的氮烯转移反应合成含氮杂环化合物的方法报道却很少.
 图式 1
金催化分子内炔烃的氮烯转移反应
Scheme1.
Examples on gold-catalyzed intramolecular nitrene transfer reaction of alkynes
图式 1
金催化分子内炔烃的氮烯转移反应
Scheme1.
Examples on gold-catalyzed intramolecular nitrene transfer reaction of alkynes
本综述将主要讨论近年来均相金催化分子间的炔烃与各种氮烯前体的反应研究进展, 与之相对的分子内的反应研究, 将不在本文的讨论范围之内.
1 金催化分子间炔烃与氮烯前体的反应
1.1 吡啶类氮叶立德作为氮烯前体
2011年, Zhang研究小组(Scheme 2)[5]和Davies研究小组(Scheme 3)[6]分别报道了金催化分子间炔烃的氮烯转移反应.相同的是, 他们两个研究小组均选用与吡啶氧化物具有类似结构的吡啶氮叶立德作为氮烯前体.不同的是, Zhang研究小组使用磺酰基取代的氮叶立德与脂肪炔烃衍生而来的炔酰胺反应, 生成α, β-不饱和脒类化合物11; 而Davies研究小组使用普通酰基取代的氮叶立德与各种炔酰胺反应, 生成多取代的噁唑类化合物.相比之下, Davies的研究工作具有更广的底物使用范围, 含有芳基、烷基、杂芳基、卤素、双键、叁键等官能团的炔酰胺都能够很好地参与反应.
 图式 2
不饱和脒的合成
Scheme2.
Synthesis of unsaturated amidine
图式 2
不饱和脒的合成
Scheme2.
Synthesis of unsaturated amidine
 图式 3
多取代噁唑的合成
Scheme3.
Synthesis of polysubstituted oxazole
图式 3
多取代噁唑的合成
Scheme3.
Synthesis of polysubstituted oxazole
Zhang的研究工作由于形成α-磺酰基亚胺金卡宾中间体后, 发生1, 2-氢迁移反应, 进而生成α, β-不饱和脒.而在Davies的研究中, 烷基取代的炔酰胺底物在反应过程中, 作者没有发现金卡宾的1, 2-氢迁移反应产物α, β-不饱和亚胺的生成, 可能是因为C—O键成键环化的速率要远远大于1, 2-氢插入金卡宾的速率.作者还提出了另一种途径:反应经过渡态E, C—O键形成与N—N键断裂协同进行, 生成中间体G, 最后释放金催化剂生成噁唑14.
在Davies研究小组的报道中, 同样的反应条件下, 芳基取代的炔醚15也可以与氮叶立德顺利反应生成噁唑17.而炔基醚的邻位有亚甲基时, 1, 2-氢迁移反应成为主要的反应途径, 生成α, β-不饱和亚胺化合物20, 没有检测到噁唑的生成(Scheme 4).
 图式 4
炔醚的氮烯转反应
Scheme4.
Nitrene transfer reaction of ynol ether
图式 4
炔醚的氮烯转反应
Scheme4.
Nitrene transfer reaction of ynol ether
之后, Davies研究小组[7].应用同样的策略实现了三芳基取代噁唑和咪唑并吡啶衍生物的合成.如Scheme 5所示, 参与反应的炔烃不局限于炔酰胺, Ar为富电子芳基的內炔也可以顺利反应得到目标产物.
 图式 5
噁唑和咪唑并吡啶的合成
Scheme5.
Oxazole and imidazo-pyridine synthesis
图式 5
噁唑和咪唑并吡啶的合成
Scheme5.
Oxazole and imidazo-pyridine synthesis
近期, Davies研究小组[8]用炔基硫醚21'与氮烯前体13在金催化下, 用邻二氯苯作溶剂、125 ℃, 环化后合成了具有高区域选择性的噁唑类化合物22.对于该反应当Ar环上连有吸电子基团时反应不发生, 而Ar环上的供电子基团会促进该反应的进行.对于不同的氮烯前体上的取代基R比如芳香基、杂芳环、胺类化合物、含醚或含醇的脂肪链都有很好的容忍度.值得一提的是该反应的区域选择性与炔酰胺相反, 可能是因为硫原子的存在, 21'与金催化剂优先形成中间体A", 再与13反应生成B", 最后发生分子内环化反应、脱去吡啶得到目标产物22(Scheme 6).
 图式 6
金催化炔基硫醚的环化反应合成噁唑类化合物
Scheme6.
Alkynyl thioethers in gold-catalyzed annulations to form oxazoles
图式 6
金催化炔基硫醚的环化反应合成噁唑类化合物
Scheme6.
Alkynyl thioethers in gold-catalyzed annulations to form oxazoles
1.2 异噁唑作为氮烯前体
2015年, 叶龙武教授课题组[9]发现异噁唑也可以作为氮烯前体, 在合适的金催化剂的作用下, 炔酰胺和它发生分子间形式上的[3+2]反应, 发展了合成多取代2-氨基吡咯的新方法.他们使用5 mol% (ArO)3PAuNTf2 (Ar=2, 4-二叔丁基苯基)作为催化剂, 1, 2-二氯乙烷(DCE)作为溶剂, 80 ℃下能够以中等至优秀的分离收率得到多取代的2-氨基吡咯化合物.有意思的是, 当使用3, 5-二取代的异噁唑(R4=H)时, 可以以100%的原子经济性得到产物4-酰基-2-氨基吡咯27; 而当异噁唑为3, 4, 5-三取代(R4≠H)时, 产物则为脱酰基的多取代2-氨基吡咯28 (Scheme 7).无论是炔酰胺或者是作为氮烯前体的异噁唑, 该反应对多种取代基团都体现了很好的兼容性.甚至使用水作溶剂, 100 ℃下也能够以良好的收率得到目标产物, 并且没有观察到炔酰胺水合产物的生成.另外, 使用该策略可以以两步、63%的分离收率合成脂肪氧合酶抑制剂的前驱体29 (Scheme 8).
 图式 7
异噁唑和炔酰胺反应合成多取代吡咯
Scheme7.
Synthesis of polysubstituted pyrrole through the reaction of isoxazoles and ynamides
图式 7
异噁唑和炔酰胺反应合成多取代吡咯
Scheme7.
Synthesis of polysubstituted pyrrole through the reaction of isoxazoles and ynamides
 图式 8
合成应用
Scheme8.
Synthetic application
图式 8
合成应用
Scheme8.
Synthetic application
至于在R4=H和R4≠H时, 会有两种不同的2-氨基吡咯产物生成, 他们也给出了较为合理的解释:当异噁唑底物为3, 4, 5-三取代(R4≠H)时, 中间体K会在反应体系中微量的水的作用下水解, 从而产生产物28.他们还结合理论计算提出了合理的反应机理(Scheme 7).
随后, 刘瑞雄课题组[10]报道了不含取代基的异噁唑与二取代丙炔酸酯的反应.在金催化剂作用下异噁唑与丙炔酸酯发生分子间的[5+2]环化后, 再进一步与另一分子异噁唑发生[4+2]环加成反应, 得到3, 8-双羰基吡啶并咪唑类化合物33.该反应对于R2是不同电子效应芳环、杂芳环(如噻吩等)、或是各种长度和大小的脂肪基团都有很好的适用性.在反应过程中, 作者还检测到了部分由氧原子进攻叁键启动的副产物吡咯的生成.根据研究结果, 作者也对该反应提出了合理的机理假设:金催化剂活化丙炔酸酯32与异噁唑(N端加成)反应, 分子内扩环得到中间体L, 再与另一份子异噁唑发生分子间的[4+2]环加成反应得到M, M经分子内开环脱水得到N, 中间体N中的吡啶环上的氮原子进攻卡宾中心碳原子得到中间体O, 释放出金催化剂得到最终产物33 (Scheme 9).
 图式 9
吡啶并咪唑和吡咯的合成
Scheme9.
Synthesis of imidazo[1, 2-a]pyridines and pyrroles
图式 9
吡啶并咪唑和吡咯的合成
Scheme9.
Synthesis of imidazo[1, 2-a]pyridines and pyrroles
2016年, Hashmi课题组[11]报道了金催化分子间的2, 1-苯并异噁唑(Anthranil) 34与炔烃发生反应, 最终以中等至优秀的收率合成了一系列的7-酰基吲哚和3-酰基喹啉衍生物. 7-酰基吲哚的形成机理如Scheme 10所示:在金催化剂的作用下, 炔烃与2, 1-苯并异噁唑反应生成中间体P, 中间体P发生分子内开环反应形成α-亚胺基金卡宾中间体Q; 然后, 分子内的C(sp2)-H插入反应生成含有吲哚骨架的中间体R; 最后释放金催化剂, 生成目标产物35.该反应具有较好的底物普适性, 能够以中等至优秀的收率得到各种7-酰基吲哚化合物, 不仅端基炔酰胺能够很好地反应(35a), 非端基炔酰胺也能够顺利地反应得到产物35b.端基炔醚、脂肪基端炔、芳基端炔以及普通內炔都可以有效地反应.当炔酰胺的炔丙位取代基有亚甲基时, 金卡宾的1, 2-H插入反应成为主要反应, 此时, 生成以较高的产率得到α, β-不饱和脒35c[11a].
 图式 10
苯并异噁唑和炔烃的反应
Scheme10.
Reaction of anthranils with alkynes
图式 10
苯并异噁唑和炔烃的反应
Scheme10.
Reaction of anthranils with alkynes
后续研究表明, 金催化2, 1-苯并异噁唑(Anthranil) 34与炔丙基醚类化合物反应, 也可以形成α-亚胺金卡宾中间体Q'. Q'经1, 2-H迁移反应形成烯基醚中间体S.此时, S很容易发生分子内的Mukaiyama-Aldol缩合反应, 并失去一分子的醇, 最终生成3-酰基喹啉衍生物(Scheme 11)[11b].与前面研究工作类似, 反应底物普适性较好, 各种供电子基团和吸电子基团、卤素、稠芳杂环取代的2, 1-苯并异噁唑都能够以较高的收率得到目标产物; 不仅活性较高的炔酰胺类炔丙基硅醚能够很好地参与反应, 普通的烷基、芳基、共轭双键取代的炔丙基硅醚也能够在三氟乙醇和1, 2-二氯乙烷作混合溶剂的条件下顺利进行反应, 以中等收率得到3-酰基喹啉衍生物37f~37h.
 图式 11
苯并异噁唑和炔丙醚的反应
Scheme11.
Reaction of anthranils with propargyl ethers
图式 11
苯并异噁唑和炔丙醚的反应
Scheme11.
Reaction of anthranils with propargyl ethers
1.3 氧氮茂作为氮烯前体
刘元红课题组[12]的研究工作表明, 二氧氮茂(dioxazole) 38也可作为氮烯前体, 在合适的金催化剂的作用下, 与炔酰胺发生分子间的反应, 生成多取代噁唑39(Scheme 12).作者认为38a、38b和38c分别在反应过程中释放出CO2、SO2和丙酮产生酰基亚胺卡宾中间体U, 中间体U中与氮原子相连的酰基氧进攻金卡宾中心碳原子, 发生分子内的关环反应, 生成中间体V, 最后, 释放催化剂得到多取代的噁唑39.然而, 研究发现38a、38b和38c在反应性上表现出较大的差别, 只有38c 能够顺利发生反应得到目标杂环产物.在最优反应条件下, 炔酰胺中的取代基R1无论是芳基、杂芳基、稠环、脂肪基还是带有双键官能团的都能够顺利发生反应.二氧氮茂38c的官能团兼容性也非常好.当R3是甲基时, 室温反应10 h, 可以以71%的收率得到噁唑39h.另外, 当R1为正丁基时, 反应5 h仅得到30%的分离收率(39c).由于没有观察到预计的金卡宾1, 2-H插入反应产物烯烃的生成, 所以, 作者认为, 该反应历程可能与Davies小组研究类似, 分子内N-酰基氧对金卡宾的亲核环化反应要远快于金卡宾的1, 2-H插入反应.之后, 万伯顺课题组[13]使用HNTf2(双三氟甲磺酰亚胺)作为Brønsted酸催化剂, 也能够催化该反应, 顺利地实现同类杂环化合物的合成.
 图式 12
金催化二氧氮茂和炔酰胺合成噁唑
Scheme12.
Gold-catalyzed synthesis of oxazole using dioxazole as nitrene precursor
图式 12
金催化二氧氮茂和炔酰胺合成噁唑
Scheme12.
Gold-catalyzed synthesis of oxazole using dioxazole as nitrene precursor
在后续研究中, 刘元红课题组[14]又报道了金催化4, 5-二氢-1, 2, 4-氧二氮茂40与炔酰胺30的反应(Scheme 13).反应温度为80 ℃, 溶剂为1, 2-二氯乙烷, 在相对温和的反应条件下实现了官能团化的4-氨基咪唑类化合物的合成.在该工作中, 杂环化合物40作为氮烯前体, 在反应过程中脱去一分子苯甲醛, 从而形成α-杂乙烯基金卡宾中间体X, 然后发生分子内关环反应, 生成中间体Y, 中间体Y释放出金催化剂后, 最终生成多取代的咪唑41.当炔酰胺30中的取代基R1为烷基时, 反应则以1, 2-氢迁移产物为主, 这也间接证明了金卡宾中间体的存在.
 图式 13
金催化4-氨基咪唑的合成
Scheme13.
Synthesis of 4-aminoimidazole via gold catalysis
图式 13
金催化4-氨基咪唑的合成
Scheme13.
Synthesis of 4-aminoimidazole via gold catalysis
最近, Hashimi课题组[15]也报道了使用1, 2, 4-氧二氮茂(Oxadiazoles) 42作为氮烯前体与炔酰胺30的反应.在金催化剂的作用下, 使用三氟甲苯作溶剂, 80 ℃发生分子间的[3+2]环化反应, 得到不同类型N-取代的4-氨基咪唑化合物43 (Scheme 14).与前面研究类似, 金催化剂首先活化炔酰胺30, 再与杂环化合物42反应生成中间体Z, Z开环后形成卡宾中间体AA, 再经分子内的环化/芳构化之后, 生成多取代咪唑43.脂肪链烃取代的炔酰胺和氧二氮茂42反应以1, 2-氢迁移的产物为主.该反应对于芳环上R1取代基的电性和位阻效应都具有较好的耐受性, 而且杂芳环噻吩也能很好地参与反应生成对应的目标产物43b.对于不同的氧二氮茂42, 在R3或者R4上增加吸电子基团时, 反应效果不太理想, 可能是因为化合物42中N原子的亲核性变弱, 从而使得整体反应效率变低, 所以即便是增加底物42的用量至5 equiv., 也只能以中等的收率得到对应的咪唑43.同样, 当R1为烷基时, 作者也能检测到部分1, 2-氢迁移产物, 这也说明反应过程中可能生成了金卡宾中间体.
 图式 14
N-酰基-4-氨基咪唑的合成
Scheme14.
Synthesis of N-acyl-4-aminoimidazole
图式 14
N-酰基-4-氨基咪唑的合成
Scheme14.
Synthesis of N-acyl-4-aminoimidazole
1.4 1, 3, 5-六氢三芳基三氰作为氮烯前体
近期, Hashmi课题组[16]报道了1, 3, 5-六氢三芳基三氰44与炔酰胺发生形式上的[2+2+2]环加成反应, 合成5-氨基四氢嘧啶45 (Scheme 15).就反应机理而言, 由44解离生成的亚胺单体44'与烯酮亚胺阳离子中间体A反应, 生成中间体AC, 中间体AC再与另一分子亚胺44'反应, 形成另一种阳离子中间体AD, 再经分子内环化反应, 生成六元环中间体AE, 最后释放出金催化剂, 并生成最终产物5-氨基四氢嘧啶类化合物45.氘代实验和竞争实验进一步验证了Scheme 15描述的反应原理, 即反应物44必须解离成亚胺单体44'之后, 才能发生形式上的[2+2+2]环加成反应.该反应底物普适性较好, 各种含供电子、缺电子的取代基的底物都可以顺利地参与反应, 以中等至优秀的收率得到目标产物.并且对于克级反应, 催化剂的负载量可降至1 mol%, 反应产率仍可以达到80%.同样, 该研究也完美地诠释原子经济性这一理念.
 图式 15
5-氨基四氢嘧啶的合成
Scheme15.
Synthesis of 5-aminotetrahydropyrimidine
图式 15
5-氨基四氢嘧啶的合成
Scheme15.
Synthesis of 5-aminotetrahydropyrimidine
1.5 氮杂环丙烯作为氮烯前体
2015年, 黄学良课题组[17]报道了一种金催化合成多取代吡咯的新方法.该方法以氮杂环丙烯49为氮烯前体, 与炔酰胺30发生分子间的形式上的[3+2]环加成反应, 在相对温和的条件下实现了多取代2-氨基吡咯50的合成(Scheme 16).该策略快速高效, 具有较好的原子经济性和底物普适性.在标准条件下, 含有不同电子效应和位阻效应取代基的芳基炔酰胺都能参与反应.杂芳环取代、脂肪取代、烯基取代的炔酰胺也都能够得到令人满意的收率.不同取代的氮杂环丙烯49也可以顺利地在反应中得到应用.值得一提的是, 当氮杂环丙烯中的R3、R4为供电子烷基取代基时, 反应变得很慢, 需要将反应温度升高至100 ℃, 可以得到相对满意的收率.根据实验结果, 作者提出了两种可能的反应机理(Scheme 16).第一种途径:中体间AF直接发生扩环反应, 生成中间体AI (path a); 另一种反应途径:反应过程中也可能生成了金卡宾中间体AG, 然后经过氮杂-4p电环化关环形成中间体AH (path b).最后中间体AI经过一系列转化得到目标产物50.
 图式 16
金催化氮杂环丙烯和炔酰胺合成2-氨基吡咯
Scheme16.
Gold-catalyzed synthesis of 2-aminoprrole through the reaction of 2H-azirines with ynamides
图式 16
金催化氮杂环丙烯和炔酰胺合成2-氨基吡咯
Scheme16.
Gold-catalyzed synthesis of 2-aminoprrole through the reaction of 2H-azirines with ynamides
在标准实验条件下, 他们还尝试使用烯基叠氮作为氮烯前体, 探究分子间有机叠氮化合物与炔酰胺的反应性.有意思的是, 烯基叠氮49a和炔酰胺30a在室温反应5 h, 仅以56%的分离收率得到多取代2-氨基吡咯50a (Eq. 1), 反应过程中并没有检测到烯基叠氮化物转化成为氮杂环丙烯49.尽管这一实验结果远没有达到理想状态, 但这代表着首例均相金催化分子间的有机叠氮化物与炔烃的反应.
1.6 叠氮化物作为氮烯前体
最近, 叶龙武课题组[18]开创性地实现了系列金催化分子间有机叠氮化物和炔酰胺的反应, 发展了合成几大类有用的含氮杂环化合物的新方法.他们发现苄基叠氮可作为氮烯前体, 参与金催化炔酰胺的氮烯转移反应.炔酰胺在含有咪唑型卡宾配体的阳离子金催化剂的作用下, 与苄基叠氮化合物反应, 脱去一分子氮气后形成α-亚胺基金卡宾中间体AK, 此时, 炔酰胺N原子上的芳基作为亲核基团与亲电性的金卡宾中间体发生C(sp2)—H键的插入反应, 形成2-氨基吲哚类化合物53.实验表明, 苄基叠氮中苯环上含有拉电子基团时(R=3-BrC6H4), 反应效果更好, 可以以较高的产率得到对应的2-氨基吲哚.当R为吲哚基团的时, 由于吲哚环较苯环具有更强的亲核性, 此时, 吲哚环优先与金卡宾中间体反应, 生成咔啉类化合物54 (Scheme 17)[18a].
 图式 17
吲哚和咔啉的合成
Scheme17.
Synthesis of indole and carboline
图式 17
吲哚和咔啉的合成
Scheme17.
Synthesis of indole and carboline
后续研究表明, 共轭的烯基炔酰胺55与苄基叠在类似的条件下, 也可以顺利参与反应.对于该反应, 作者提出了氮杂-Nazorov环化反应机理(Scheme 18).通过该反应, 作者可以快捷、高效地合成一系列多取代吡咯杂环化合物[18b].
 图式 18
叠氮化物和炔酰胺的反应合成吡咯化合物
Scheme18.
Synthesis of pyrrole via the reaction of azides and ynamides
图式 18
叠氮化物和炔酰胺的反应合成吡咯化合物
Scheme18.
Synthesis of pyrrole via the reaction of azides and ynamides
而对于含有供电子基团的苄基叠氮(如: R=4-MeC6H4)作为氮烯前体, 反应只能以30%的收率得到相应的2-氨基吲哚.除此之外, 他们还观察到了另一种氢迁移产物氮杂-1, 3-丁二烯的生成.通过调节炔酰胺中与氮原子相连的苯环上R2基团的电性, 他们可以以96%的分离产率得到58, 双键的顺、反比例最高可以超过20:1.结合理论计算, 他们认为该反应在生成金卡宾中间体后, 极有可能经历了去质子化和质子去金化这一协同反应历程(AV→AW, Scheme 19)[18c].当R1含有亚甲基时, 生成中间体AV'直接发生1, 2-氢迁移从而得到α, β-不饱和脒类化合物[18d].
 图式 19
金催化合成2-杂-1, 3-丁二烯
Scheme19.
Gold-catalyzed synthesis of 2-aza-1, 3-butadiene
图式 19
金催化合成2-杂-1, 3-丁二烯
Scheme19.
Gold-catalyzed synthesis of 2-aza-1, 3-butadiene
2015年, 黄学良课题组[19]和刘瑞雄课题组[20]几乎同时完成了金催化分子间烯基叠氮化物和炔酰胺的反应研究.在他们各自的条件下均可以得到[3+2]环加成产物2-氨基吡咯化合物和[3+4]环加成产物一氢苯并[d]氮杂环庚三烯化合物.研究发现, 反应物结构对反应类型有较大的影响.比如:芳环间位含有强供电子基团(甲氧基)的炔酰胺与烯基叠氮化物反应主要得到形式上[3+4]环加成产物; 普通非间位供电子取代的炔酰胺和烯基叠氮化物反应, 则得到的是形式上[3+2]环加成的主产物.从七元环产物的结构可知, 反应过程中, 烯基叠氮59极有可能原位生成氮杂环丙烯49, 再发生后续的环化反应(Scheme 20).该反应具有较好底物普适性, 能够以较高的收率得到相应的多取代吡咯杂环化合物.
 图式 20
烯基叠氮化物和炔酰胺的反应
Scheme20.
Reaction of vinyl azides with ynamides
图式 20
烯基叠氮化物和炔酰胺的反应
Scheme20.
Reaction of vinyl azides with ynamides
通过1, 2-氢迁移反应产生α, β-不饱和双键是验证反应过程是否存在卡宾机理重要方法之一.基于此, 黄学良课题组发现对于含有烷基基团的炔酰胺30d, 反应不只生成吡咯64, 还生成了二氢吡啶65.向反应体系中加3 equiv.的重水, 他们发现化合物65中含有部分的氘原子(小于15%).根据这一实验结果, 他们提出了如Scheme 21所示的两种可能的反应路径.
 图式 21
机理实验
Scheme21.
Mechanistic studies
图式 21
机理实验
Scheme21.
Mechanistic studies
最近, 黄学良课题组[21]又尝试使用叠氮化合物69作为氮烯前体, 检验它与炔酰胺30b在金催化剂作用下的反应活性.实验结果表明, 反应过程中并没有预计产物72生成, 有机叠氮69大部分转化为吡啶并吲唑70a.同时, 他们分离得到了少量3-氨基吲哚类化合物71 (Scheme 22), 其结构通过晶体解析得到进一步确证.对照实验表明, 在类似的反应条件下, 70a和30b反应可以以较高产率得到多取代吲哚71, 且反应过程并没有检测到71的区域异构体2-氨基吲哚的生成.这一结果表明, 在金催化剂的作用下, 70a对30b亲核加成反应选择性的发生在炔酰胺中氮原子的β位[20].
 图式 22
叠氮化物69和炔酰胺30b的反应
Scheme22.
Reaction of azides 69 with ynamide 30b
图式 22
叠氮化物69和炔酰胺30b的反应
Scheme22.
Reaction of azides 69 with ynamide 30b
从前大部分已报道的工作可以看出, 炔酰胺在阳离子型金催化剂的作用下, 可以生成烯酮亚胺阳离子中间体, 此时, 氮原子的α位具有很强的亲电性, 因此, 亲核加成应该优先发生在氮原子的α位.为了得到更多有关反应机理方面的信息, 他们合成了金催化剂与炔酰胺的配合物, 并且成功地培养了该配合物的晶体.根据晶体结构解析结果可以清楚的发现, 炔酰胺30b在金催化剂的作用下发生了分子内环化反应, 生成了一个新的五元环(图 2).
 图2
JohnphosAu(30b)SbF6的单晶结构
Figure2.
X-Ray structure of the complex JohnphosAu-(30b)SbF6
图2
JohnphosAu(30b)SbF6的单晶结构
Figure2.
X-Ray structure of the complex JohnphosAu-(30b)SbF6
这一实验结果很好地解释了文章中报道的不同寻常的区域选择性(β位选择性).他们认为, 金催化剂与炔酰胺反应生成阳离子中间体AV, 此时, 氮原子的β位碳原子更具亲电性, 因此, 70对中间体AV的开环反应优先发生在β位的碳原子上, 进而生成中间体AW.中间体AW发生开环反应后生成中间体AX.动力学实验表明, 吡啶并吲唑70中a环上取代基的电子效应对反应的速率有较大影响.大体规律是X为吸电子基团时的反应速度要大于X为给电子基团时的反应速度.分子内的傅-克烷基化或者卡宾的C-H插入反应机理都不能解释这一实验现象.因此, 作者提出了类似于苯环Csp2—H键活化的反应机理.最终中间体AX发生分子内环化, 再经异构化生成吲哚71 (Scheme 23).
 图式 23
3-氨基吲哚类化合物的合成
Scheme23.
Synthesis of 3-amido-indole
图式 23
3-氨基吲哚类化合物的合成
Scheme23.
Synthesis of 3-amido-indole
2 结论与展望
金催化分子间氮烯前体与炔烃的反应为合成含有不同结构类型的氮杂环化合物提供了多种便捷、有效的方法.对于分子内炔烃的氮烯转移反应来说, 底物来源受限非常明显, 尤其在需要建立杂环化合物库的时候, 不同官能化的化合物需要相对应的预官能化底物.这些不同官能化的底物的合成就严重制约了分子内反应发展.而分子间反应在扩充化合物库的时候优点就再明显不过了, 比如各种官能化的炔烃都可以直接购买, 或者通过简单的方法合成得到.不同官能化的氮烯前体也相对容易得到.在合适的条件下, 通过分子间反应交叉组合起来, 有望合成得到一个相对庞大的化合物库.目前为止, 有关金催化分子间炔烃的氮烯转移反应尚处于起步阶段.虽然, 它在高效合成各种官能团化的含氮杂环化合物方面已经展现出优越性, 但是, 该类反应在底物类型方面还存在明显的局限性.如:涉及的炔烃大多局限于炔酰胺, 有关普通的非极化炔烃的成功例子报道较少; 新型的氮烯前体还有待科研工作者们去继续发现、发掘、创造和利用.另外, 很多含有氮杂环的天然产物和具有生物、药物活性的化合物都是手性化合物, 而现在已报道的金催化分子间氮烯转移和炔烃的研究, 都还没有涉及立体中心、手性合成方面的工作.与此相关的研究工作也有待有机化学工作者去继续发掘开拓.
-
-
[1]
(a) Pozharskii, A. F.; Soldatenkov, A. T.; Katritzky, A. R. Heterocycles in Life and Society, Wiley, New York, 1997.
(b) Katritzky, A. R. Handbook of Heterocyclic Chemistry, Per-gamon Press, Oxford, 1985.
(c) Balaban, A. T.; Oniciu, D. C.; Katritzky, A. R. Chem. Rev. 2004, 104, 2777. -
[2]
Related reviews, see:(a) Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410.
(b) Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180.
(c) Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351.
(d) Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326.
(e) Fürstner, A. Chem. Soc. Rev. 2009, 38, 3208.
(f) Hashmi, A. S. K. Angew. Chem., Int. Ed.2010, 49, 5232.
(g) Xiao, J.; Li, X. Angew. Chem., Int. Ed. 2011, 50, 7226.
(h) Friend, C. M.; Hashmi, A. S. K. Acc. Chem. Res. 2014, 47, 729.
(i) Zhang, L. Acc. Chem. Res. 2014, 47, 877.
(j) Yeom, H.-S.; Shin, S. Acc. Chem. Res. 2014, 47, 966.
(k) Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028.
(l) Qian, D.; Zhang, J. Chem. Soc. Rev. 2015, 44, 677.
(m) Davies, P. W.; Garzûn, M. Asian J. Org. Chem. 2015, 4, 694.
(n) Huple, D. B.; Ghorpade, S.; Liu, R.-S. Adv. Synth. Catal. 2016, 358, 1348.
(o) Pan, F.; Shu, C.; Ye, L.-W. Org. Biomol. Chem. 2016, 14, 9456.
(p) Shu, C.; Li, L.; Tan, T.-D.; Yuan, D.-Q.; Ye, L.-W. Sci. Bull. 2017, 62, 352. -
[3]
Recent related reviews, see:(a) Davies, P. W.; Garzûn, M. Asian J. Org. Chem. 2015, 4, 694.
(b) Song, X.-R.; Qiu, Y.-F.; Liu, X.-Y.; Liang, Y.-M. Org. Bio-mol. Chem. 2016, 14, 11317. -
[4]
(a) Gorin, D. J.; Davis, N. R.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 11260.
(b) Huo, Z.; Yamamoto, Y. Tetrahedron Lett. 2009, 50, 3651.
(c) Wetzel, A.; Gagosz, F. Angew. Chem., Int. Ed. 2011, 50, 7354.
(d) Lu, B.; Luo, Y.; Liu, L.; Ye, L.; Wang, Y.; Zhang, L. Angew. Chem., Int. Ed. 2011, 50, 8358.
(e) Yan, Z.-Y.; Xiao, Y.; Zhang, L. Angew. Chem., Int. Ed. 2012, 51, 8624.
(f) Xiao, Y.; Zhang, L. Org. Lett. 2012, 14, 4662.
(g) Gronnier, C.; Boissonnat G.; Gagosz, F. Org. Lett. 2013, 15, 4234.
(h) Prechter, A.; Henrion, G.; Bel, P. F.; Gagosz, F. Angew. Chem., Int. Ed. 2014, 53, 4959.
(i) Shen, C.-H.; Pan, Y.; Yu, Y.-F.; Wang, Z.-S.; He, W.; Li, T.; Ye, L.-W. J. Organomet. Chem. 2015, 795, 63.
(j) Li, N.; Wang, T. Y.; Gong, L. Z.; Zhang, L. Chem. Eur. J. 2015, 21, 3585.
(k) Pan, Y.; Chen, G.-W.; Shen, C.-H.; He, W.; Ye, L.-W. Org. Chem. Front. 2016, 3, 491.
(l) Li, N.; Lian, X. L.; Li, Y. H.; Wang, T. Y.; Han, Z. Y.; Zhang, L.; Gong, L. Z. Org. Lett. 2016, 18, 4178.
(m) Lonca, G. H.; Tejo, C.; Chan, H. L.; Chiba, S.; Gagosz, F. Chem. Commun. 2017, 53, 736.
(n) Zhang, X.-X.; Sun, X.-P.; Fan, H.; Li, P.; Lyu, C.; Rao, W.-D. Eur. J. Org. Chem. 2016, 25, 4265.
(o) Zhang, X.-X.; Sun, X.-P.; Fan, H.; Lyu, C.; Li, P.; Zhang, H.-F.; Rao, W.-D. RSC Adv. 2016, 6, 56319.
(p) Zhang, X.-X; Li, P.; Lyu, C.; Yong, W.-X.; Li, J.; Zhu, X.-B.; Rao, W.-D. Org. Biomol. Chem. 2017, 15, 6080.
(q) González, J.; Santamaría, J.; Suárez-Sobrino, Á. L.; Ballesteros, A. Adv. Synth. Catal. 2016, 358, 1398. -
[5]
Li, C.; Zhang, L. Org. Lett. 2011, 13, 1738. doi: 10.1021/ol2002607
-
[6]
(a) Davies, P. W.; Cremonesi, A.; Dumitrescu, L. Angew. Chem., Int. Ed. 2011, 50, 8931.
(b) Gillie, A. D.; Reddy, R. J.; Davies, P. W. Adv. Synth. Catal. 2016, 358, 226. -
[7]
(a) Chatzopoulou, E.; Davies, P. W. Chem. Commun. 2013, 49, 8617.
(b) Garzûn, M.; Davies, P. W. Org. Lett. 2014, 16, 4850. -
[8]
Reddy, R. J.; Ball-Jones, M. P.; Davies, P. W. Angew. Chem., Int. Ed. 2017, 56, 13310.. doi: 10.1002/anie.201706850
-
[9]
(a) Zhou, A. -H. ; He, Q. ; Shu, C. ; Yu, Y. -F. ; Liu, S. ; Zhao, T. ; Zhang, W. ; Lu, X. ; Ye, L. -W. Chem. Sci. 2015, 6, 1265.
(b) Xiao, X. -Y. ; Zhou, A. -H. ; Shu, C. ; Pan, F. ; Li, T. ; Ye, L. -W. Chem. Asian J. 2015, 10, 1854.
(c) Li, X. -L. ; Wang, J. -Q. ; Li, L. ; Yin, Y. -W. ; Ye, L. -W. Acta Chim. Sinica 2016, 74, 49(in Chinese).
(李新玲, 王佳琪, 李龙, 尹应武, 叶龙武, 化学学报, 2016, 74, 49. )
(d) Shen, W. -B. ; Xiao, X. -Y. ; Sun, Q. ; Zhou, B. ; Zhu, X. -Q. ; Yan, J. -Z. ; Lu, X. ; Ye, L. -W. Angew. Chem., Int. Ed. 2017, 56, 605. -
[10]
Sahani, R. L.; Liu, R.-S. Angew. Chem., Int. Ed. 2017, 56, 1026. doi: 10.1002/anie.201610665
-
[11]
(a) Jin, H.; Huang, L.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2016, 55, 794.
(b) Jin, H.; Tian, B.; Song, X.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2016, 55, 12688.
(c) Sahani, L.-R.; Liu, R.-S. Angew. Chem., Int. Ed. 2017, 56, 12736. -
[12]
Chen, M.; Sun, N.; Chen, H.-Y.; Liu, Y.-H. Chem. Commun. 2016, 52, 6324. doi: 10.1039/C6CC02776H
-
[13]
Zhao, Y.-Y.; Hu, Y.-C.; Wang, C.-X.; Li, X.-C.; Wan, B.-S. J. Org. Chem. 2017, 82, 3935. doi: 10.1021/acs.joc.7b00076
-
[14]
Xu, W.; Wang, G.-N.; Sun, N.; Liu, Y.-H. Org. Lett. 2017, 19, 3307. doi: 10.1021/acs.orglett.7b01469
-
[15]
Zeng, Z.-Y.; Jin, H.-M.; Xie, J.; Tian, B.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Org. Lett. 2017, 19, 1020. doi: 10.1021/acs.orglett.7b00001
-
[16]
Zeng, Z.; Jin, H.; Song, X.; Wang, Q.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Chem. Commun. 2017, 53, 4304. doi: 10.1039/C7CC00789B
-
[17]
Zhu, L.; Yu, Y.; Mao, Z.; Huang, X. Org. Lett. 2015, 17, 30. doi: 10.1021/ol503172h
-
[18]
(a) Shu, C.; Wang, Y.-H.; Zhou, B.; Li, X.-L.; Ping, Y.-F.; Lu, X.; Ye, L.-W. J. Am. Chem. Soc. 2015, 137, 9567.
(b) Shu, C.; Wang, Y.-H.; Shen, C.-H.; Ruan, P.-P.; Lu, X.; Ye, L.-W. Org. Lett. 2016, 18, 3254.
(c) Shu, C.; Shen, C.-H.; Wang, Y.-H.; Li, L.; Li, T.; Lu, X.; Ye, L.-W. Org. Lett. 2016, 18, 4630.
(d) Ruan, P.-P.; Li, H.-H.; Liu, X.; Zhang, T.; Zuo, S.-X.; Zhu, C.-Y.; Ye, L.-W. J. Org. Chem. 2017, 82, 9119. -
[19]
Wu, Y.; Zhu, L.; Yu, Y.; Luo, X.; Huang, X. J. Org. Chem. 2015, 80, 11407. doi: 10.1021/acs.joc.5b02057
-
[20]
Pawar, S. K.; Sahani, R. L.; Liu, R.-S. Chem. Eur. J. 2015, 21, 10843. doi: 10.1002/chem.v21.30
-
[21]
Yu, Y.; Chen, G.; Zhu, L.; Liao, Y.; Wu, Y.; Huang, X. J. Org. Chem. 2016, 81, 8142. doi: 10.1021/acs.joc.6b01948
-
[1]
-

图 1 生命体中的含氮杂环化合物
Figure 1 Selected examples on N-heterocyclic compounds in living organisms

图式 1 金催化分子内炔烃的氮烯转移反应
Scheme 1 Examples on gold-catalyzed intramolecular nitrene transfer reaction of alkynes

图式 6 金催化炔基硫醚的环化反应合成噁唑类化合物
Scheme 6 Alkynyl thioethers in gold-catalyzed annulations to form oxazoles

图式 7 异噁唑和炔酰胺反应合成多取代吡咯
Scheme 7 Synthesis of polysubstituted pyrrole through the reaction of isoxazoles and ynamides

图式 12 金催化二氧氮茂和炔酰胺合成噁唑
Scheme 12 Gold-catalyzed synthesis of oxazole using dioxazole as nitrene precursor

图式 16 金催化氮杂环丙烯和炔酰胺合成2-氨基吡咯
Scheme 16 Gold-catalyzed synthesis of 2-aminoprrole through the reaction of 2H-azirines with ynamides

图式 18 叠氮化物和炔酰胺的反应合成吡咯化合物
Scheme 18 Synthesis of pyrrole via the reaction of azides and ynamides

图 2 JohnphosAu(30b)SbF6的单晶结构
Figure 2 X-Ray structure of the complex JohnphosAu-(30b)SbF6
-


 下载:
下载:
























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 19
- 文章访问数: 5517
- HTML全文浏览量: 396
 下载:
下载:
