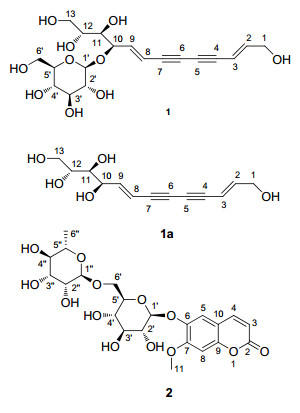
 图1
化合物1, 1a和2的结构
Figure1.
Structures of compounds 1, 1a, and 2
图1
化合物1, 1a和2的结构
Figure1.
Structures of compounds 1, 1a, and 2
引用本文:
绪扩, 冯子明, 杨桠楠, 姜建双, 张培成. 茅苍术根茎中的两个新化合物[J]. 有机化学,
2017, 37(11): 3019-3023.
doi:
10.6023/cjoc201706031

Citation: Xu Kuo, Feng Ziming, Yang Ya'nan, Jiang Jianshuang, Zhang Peicheng. Two New Compounds from Rhizomes of Atractylodes lancea[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 3019-3023. doi: 10.6023/cjoc201706031
Citation: Xu Kuo, Feng Ziming, Yang Ya'nan, Jiang Jianshuang, Zhang Peicheng. Two New Compounds from Rhizomes of Atractylodes lancea[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 3019-3023. doi: 10.6023/cjoc201706031
茅苍术根茎中的两个新化合物
摘要:
运用多种柱色谱技术对茅苍术根茎进行了系统的化学成分研究,从其80%乙醇提取物中分离得到一个新的4,6-二炔烯类化合物和一个新的香豆素类化合物,分别命名为(10R,11R,12R)-十三烷-2E,8E-二烯-4,6-二炔-1,10,11,12,13-五醇-10-O-β-D-吡喃葡萄糖苷(1)和isoscopoletin 6-O-α-L-吡喃鼠李糖基-(1→6)-β-D-吡喃葡萄糖苷(2).其平面结构通过UV,IR,HR-ESIMS,1D和2D NMR等谱学技术确定.化合物1的相对构型通过纽曼投影式分析优势构象并结合1D NOESY的方法确定,而绝对构型通过比较电子圆二色谱(ECD)的方法确定.相关结果为链状多元醇的立体构型研究提供了参考.在10 μmol·L-1浓度下,化合物1和2对α-葡萄糖苷酶及蛋白酪氨酸磷酸酶1B表现出弱的抑制活性.
English
Two New Compounds from Rhizomes of Atractylodes lancea
Abstract:
In our phytochemical investigations on the 80% EtOH extract of the rhizomes of Atractylodes lancea, two new compounds named (10R, 11R, 12R)-tridecane-2E, 8E-diene-4, 6-diyne-1, 10, 11, 12, 13-pentol-10-O-β-D-glucopyranoside (1) and isoscopoletin 6-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside (2) were isolated using various column chromatographic methods. Their planar structures were elucidated by means of spectroscopic and spectrometric analyses (UV, IR, 1D and 2D NMR, and HR-ESIMS). The relative configurational assignment of 1 was deduced by visualized Newman projections in association with the 1D NOESY spectra, whereas its absolute configuration was established by comparison of experimental electronic circular dichroism (ECD) spectra with known compounds. These results will benefit subsequent stereochemical studies of those open-chain polyhydric alcohols. Compounds 1 and 2 showed weak inhibitory effects on α-glucosidases and protein tyrosine phosphates 1B (PTP1B) at a concentration of 10 μmol·L-1.
-
Key words:
- Atractylodes lancea
- / 4, 6-diyne
- / polyacetylene
- / coumarin
- / absolute configuration
-
苍术为菊科苍术属Atractylodes, 多年生草本植物, 包括7个植物种群, 主要分布于亚洲东部, 其中, 我国特有5个种群[1].苍术属植物的药用历史最早记载于2000多年前的《神农本草经》, 以苍术和白术最为常用.药用苍术为茅苍术(Atractylodes lancea)和关苍术(Atractylodes chinensis)的干燥根茎, 具有燥湿健脾, 除风散寒、明目之功效[2].现代药理学研究表明, 苍术具有促进胃肠蠕动、利尿、肝保护、抗炎、抗病毒、降血脂、降血糖等方面的药理作用[3, 4].其主要含有萜类、多烯炔类、甾体类、酰基蔗糖类以及其它类化学成分[5~10].本工作对茅苍术80%乙醇提取物的水溶性化学成分进行了研究, 从中分离得到2个新化合物(图 1).借助UV, IR, HR-ESIMS, 1D和2D NMR以及ECD等多种谱学技术, 鉴定其结构分别为(10R, 11R, 12R)-十三烷-2E, 8E-二烯-4, 6-二炔-1, 10, 11, 12, 13-五醇-10-O-β-D-吡喃葡萄糖苷(1)和isoscopoletin 6-O-α-L-吡喃鼠李糖基-(1→6)-β-D-吡喃葡萄糖苷(2).还评价了化合物1和2对α-葡萄糖苷酶以及蛋白酪氨酸磷酸酶1B (PTP1B)的体外抑制活性.
图1
化合物1, 1a和2的结构
Figure1.
Structures of compounds 1, 1a, and 2
1 结果与讨论
1.1 化合物1的结构鉴定
化合物1为棕色无定型粉末, 易溶于水和甲醇; [α]D20-136.0 (c 0.12, MeOH); UV-Vis (MeOH) λmax [log ε/(L•mol-1•cm-1)]: 216 (4.46), 263 (3.95), 277 (4.19), 294 (4.35), 314 (4.27) nm; ECD (MeOH) λmax [Δε/(L•mol-1•cm-1)]: 233 (-5.43), 293 (-1.50), 312 (-1.03) nm; 其HR-ESIMS和13C NMR谱提示分子式为C19H26O10 (calcd for C19H25O10 [M-H]- 413.1448, found 413.1452), 不饱和度为7.该化合物的红外光谱提示结构中存在羟基(3352 cm−1)、炔键(2133, 2208 cm−1)和烯键(1369, 1417, 1628 cm−1).极具特征的紫外图谱提示其为烯炔类衍生物, 且具有丁二炔基与烯键共轭形成的发色团(CH=CHC≡CC≡CCH=CH)[11, 12].结合HSQC二维谱, 1H NMR谱(表 1)显示1组Δ2, 3烯键信号δH 5.86, 6.49, 其3J值(16.0 Hz)提示其为反式构型; 1组Δ8, 9烯键信号δH 6.20, 6.38, 其3J值(16.0 Hz)提示其为反式构型; 1组糖基氢信号, 其端基的3J值为7.5 Hz; 3组含氧次甲基的氢信号δH 3.29~3.34, 3.51~3.61, 4.52; 2组含氧亚甲基的氢信号δH 3.35~3.43 (1H), 3.51~3.61 (1H), 4.06 (2H). 13C NMR谱(表 1)中可推断出4个烯键碳信号δC 106.2, 109.3, 147.2, 149.4; 4个响应较低的炔碳信号δC 80.6, 73.5, 74.0, 80.6; 3个含氧次甲基碳信号δC 70.8, 73.1, 77.3; 2个含氧亚甲基碳信号δC 60.8, 63.1以及1组吡喃葡萄糖基信号δC 101.8, 73.5, 76.7, 70.6, 76.6, 61.5, 其相对构型由端基氢的3J值(7.5 Hz)确定为β构型(葡萄糖基的绝对构型通过气相色谱法确定为D). 1H-1HCOSY谱(图 2)中发现H2-1/H-2, H-2/H-3之间以及H-8/H-9, H-9/H-10, H-10/H-11, H-11/H-12, H-12/H2-13之间存在相关, 确定结构中存在C1(H2)OHC2(H)=C3(H)和C8(H)=C9(H)C10(H)ORC11(H)OHC12(H)OHC13(H2)OH的片段. HMBC谱(图 2)中发现H-1与C-3相关, H-8与C-6相关, H-9与C-7相关, 提示1为4, 6-二炔烯苷类, 进一步分析发现葡萄糖的端基氢H-1′与C-10存在相关.因此, 化合物1的平面结构被确定为十三烷-2E, 8E-二烯-4, 6-二炔-1, 10, 11, 12, 13-五醇-10-O-β-D-吡喃葡萄糖苷.
 表 1
化合物1和2的1H和13C NMR数据(DMSO-d6)
Table 1.
1H NMR spectroscopic data for compounds 1 and 2 in DMSO-d6
表 1
化合物1和2的1H和13C NMR数据(DMSO-d6)
Table 1.
1H NMR spectroscopic data for compounds 1 and 2 in DMSO-d6
No. 1a 1ab No. 2a δH (J in Hz) δC δH (J in Hz) δC δH (J in Hz) δC 1 4.06, brs 60.8 4.11, dd (2.0, 4.5) 62.7 2 160.5 2 6.49, dt (16.0, 4.5) 149.4 6.35, dt (16.0, 4.5) 147.7 3 6.26, d (9.5) 113.3 3 5.86, d (16.0) 106.2 5.81, brd (16.0) 108.8 4 7.86, d (9.5) 144.4 4 80.6 80.4 5 7.27, s 113.0 5 73.5 74.8 6 143.2 6 74.0 74.7 7 152.9 7 80.6 80.6 8 7.10, s 100.4 8 6.20, d (16.0) 109.3 5.88, d (16.0) 109.9 9 150.1 9 6.38, dd (16.0, 5.0) 147.2 6.40, dd (16.0, 5.0) 149.4 10 111.2 10 4.52, dd (5.0, 2.5) 77.3 4.41, dt (5.0, 2.5) 72.2 11 3.87, s 56.3 11 3.29~3.34, m 73.1 3.42, dd (8.0, 2.5) 74.6 1′ 4.91, d (7.5) 100.4 12 3.51~3.61, m 70.8 3.61~3.66, m 72.9 2′ 3.26~3.29, m 73.1 13a 3.51~3.61, m 63.1 3.57, dd (11.5, 5.0) 64.9 3′ 3.23~3.26, m 76.8 13b 3.35~3.43, m 3.74, dd (11.5, 5.0) 4′ 3.06~3.12, m 69.9 1′ 4.08, d (7.5) 101.8 5′ 3.48~3.51, m 75.6 2′ 3.02~3.07, m 73.5 6′a 3.84, d (11.5) 66.4 3′ 3.09~3.12, m 76.7 6′b 3.37~3.40, m 4′ 2.95~3.01, m 70.6 1'' 4.51, d (1.0) 100.5 5′ 3.13~3.17, m 76.6 2'' 3.50~3.53, m 70.4 6′a 3.69, brd (11.5) 61.5 3'' 3.45, dd (9.0, 3.0) 70.7 6′b 3.35~3.43, m 4'' 3.13~3.19, m 72.0 5'' 3.39~3.42, m 68.4 6'' 1.08, d (6.0) 17.9 a在DMSO-d6溶液中测得的数据; b在MeOH-d4甲醇溶液中测得的数据.  图2
化合物1和2的HMBC和1H-1HCOSY二维谱的关键相关信号
Figure2.
Key HMBC and 1H-1HCOSY correlations of 1 and 2
图2
化合物1和2的HMBC和1H-1HCOSY二维谱的关键相关信号
Figure2.
Key HMBC and 1H-1HCOSY correlations of 1 and 2
为确定该结构的立体构型, 将化合物1在温和条件下用蜗牛酶进行水解, 得到其苷元1a(图 1).通过纽曼投影式对化合物1a进行构象分析[13], 分别以C10-C11和C11-C12为轴进行投影, 可得到12种构象(图 3).化合物1a的1H NMR谱中显示3J10, 11, 3J11, 12值分别为2.5, 8.0 Hz (测试溶剂: MeOH-d4), 说明H-10/H-11和H-11/H-12分别为邻位交叉式(红色标记)和对位交叉式(紫色标记)构象.化合物1a的1D NOESY显示H-11与H-9相关, H-12与H-10相关, 而未发现H-13和H-10的相关信号, 提示化合物1a中C10-C11与C11-C12的相对构型分别为苏式和赤式(图 3).在甲醇溶液中测得化合物1的ECD谱图(图 4), 通过比较1和已知化合物[(10R, 11R)-, (10R, 11S)-, (10S, 11R)-十三烷-2E, 8E, 12-三烯-4, 6-二炔-1, 10, 11-三醇-10-O-β-D-吡喃葡萄糖苷]的ECD谱图, 得知化合物在200~400 nm范围内的Cotton效应仅体现C-10的手性, 进一步说明C-10的构型为R[14].综上所述, 化合物1的结构最终被确定为(10R, 11R, 12R)-十三烷-2E, 8E-二烯-4, 6-二炔-1, 10, 11, 12, 13-五醇-10-O-β-D-吡喃葡萄糖苷.
 图3
化合物1a的纽曼投影式和1D NOESY相关
Figure3.
Newman projections and 1D NOESY correlations of compound 1a
图3
化合物1a的纽曼投影式和1D NOESY相关
Figure3.
Newman projections and 1D NOESY correlations of compound 1a
1.2 化合物2的结构鉴定
化合物2为白色无定型粉末, 易溶于甲醇、水; [α]D20-82.5 (c 0.10, MeOH), UV-Vis (MeOH) λmax [log ε/(L•mol-1•cm-1)]: 224 (4.56), 255 (3.86), 287 (3.99), 336 (4.14) nm; 其HR-ESIMS和13C NMR谱提示分子量为C22H28O13 (calcd for C22H28O13Na [M+Na]+ 523.1428, found 523.1438), 不饱和度为9.该化合物的红外光谱提示结构中存在羟基(3406 cm−1)、共轭羰基(1710 cm−1)和苯环(1616, 1515 cm−1).结合HSQC二维谱, 1H NMR谱(表 1)显示1组Δ3, 4烯键信号δH 6.26, 7.86, 其3J值(9.5 Hz)提示为顺式构型; 2个孤立的芳香氢信号δH 7.10, 7.27; 1组甲氧基氢信号δH 3.87; 2组糖取代基氢信号, 其端基的3J值为1.0和7.5 Hz. 13C NMR谱(表 1)中可推断出8个芳香碳信号δC 113.3, 144.4, 113.0, 143.2, 152.9, 100.4, 150.1, 111.2; 1个共轭羰基碳信号δC 160.5; 1组吡喃葡萄糖基信号δC 100.4, 73.1, 76.8, 69.9, 75.6, 66.4和1组吡喃鼠李糖基信号δC 100.5, 70.4, 70.7, 72.0, 68.4, 17.9, 其相对构型由端基氢的3J值分别确定为α和β构型(葡萄糖基和鼠李糖基的绝对构型通过气相色谱法分别确定为D和L).除糖取代基和甲氧基信号外, 碳谱中的8个芳香碳信号和1个共轭羰基碳信号, 提示结构中存在1个C6-C3单元, 该化合物为香豆素母核[15]; 结合氢谱中的信号, 进一步推测该化合物为6, 7-二连氧取代型的香豆素类骨架. HMBC二维谱(图 2)显示, 甲氧基氢(H3-11)与C-7存在相关, 葡萄糖基的H-1'与C-6存在相关, 鼠李糖基的H-1''与葡萄糖基的C-6存在相关.确证化合物2的结构为isoscopoletin 6-O-α-L-吡喃鼠李糖基-(1→6)-β-D-吡喃葡萄糖苷.
 图4
化合物1与模型化合物的实验ECD谱图
Figure4.
Experimental ECD spectra of 1 and model compounds
图4
化合物1与模型化合物的实验ECD谱图
Figure4.
Experimental ECD spectra of 1 and model compounds
1.3 体外药理活性筛选
化合物1和2对α-葡萄糖苷酶以及蛋白酪氨酸磷酸酶1B (PTP1B)表现出弱的抑制活性.在10 µmol•L-1的浓度下, 化合物1和2对α-葡萄糖苷酶的体外抑制活性分别为20.4%和9.7%;对PTP1B的体外抑制活性分别为3.0%和18.0%.
2 实验部分
2.1 仪器与试剂
JASCO P-2000型旋光光度仪; JASCO V-650型紫外可见分光光度计; JASCO J-815型圆二色光谱仪; Nicolet 5700型红外光谱分析仪; Bruker AVANCE Ⅲ 500型和Agilent/Varian Directdrive2 500型核磁共振仪(残留溶剂峰为内标); Agilent 6520型高分辨质谱仪; Agilent 1260型高效液相色谱仪; Shimadzu LC-10AT制备型高效液相色谱仪; Rp-18型十八烷基键合反相硅胶(50 μm, 日本YMC公司); LH-20型葡聚糖凝胶(瑞典Pharmacia精细化工); HP-20型大孔吸附树脂(日本三菱化学公司); ODS反相制备色谱柱(25 cm×20 mm, 5 μm, 日本YMC公司); Apollo C18分析色谱柱(25 cm×4.6 mm, 5 μm).
2.2 实验样品
茅苍术根茎于2014年6月购自湖北省黄冈市, 经中国医学科学院(北京协和医学院)药物研究所的马林老师鉴定为Atractylodes lancea.该植物标本(ID-s-2596)现存放于中国医学科学院(北京协和医学院)药物研究所标本室.
2.3 提取与分离
将100 kg干燥的茅苍术根茎粉碎, 80%乙醇浸泡12 h, 然后在85 ℃下回流提取(150 L×3), 每次提取2 h.合并后的提取液经减压浓缩后得25.6 kg浸膏.用50 L蒸馏水将浸膏充分分散, 依次用石油醚、乙酸乙酯和正丁醇萃取.正丁醇萃取液经减压浓缩、冷冻干燥后得1.2 kg萃取物.用1.5 L蒸馏水超声溶解正丁醇萃取物, 过滤后经HP-20型大孔吸附树脂柱(120 cm×15 cm)进行梯度洗脱, 洗脱溶剂依次为蒸馏水、15%乙醇、30%乙醇、50%乙醇及95%乙醇.各梯度溶液洗脱40 L, 合并流份后减压回收溶剂, 得水部位824.0 g (A), 15%乙醇亚部位88.6 g (B), 30%乙醇亚部位106.4 g (C), 50%乙醇亚部位53.3 g (D), 95%乙醇亚部位19.5 g (E).选择30%乙醇亚部位(96.4 g)进行Rp-18型反相硅胶柱(50 cm×8 cm, 50 μm)的中压梯度洗脱, 洗脱液为0~100%甲醇溶液, 每个梯度洗脱约4 L流动相.按照每500 mL为1瓶流份收集样品, 根据HPLC-DAD检测结果合并流份, 得10个亚部位(Fr.C1~Fr.C10). Fr.C1亚部位(30.5 g)经LH-20型葡聚糖凝胶柱(80 cm×6 cm)进行洗脱, 洗脱液为蒸馏水, 共得到123瓶流份(100 mL/瓶, Fr.C1.1~Fr.C1.123).各组分经HPLC-DAD分析后, 减压回收溶剂、冷冻干燥得干燥固体. Fr.C1.49~Fr.C1.56部位直接经C18反相制备液相色谱柱(250 mm×20 mm, 5 μm)纯化[V(甲醇): V(水)=20: 80]后得化合物1 (22.9 mg). Fr.C1.(57~95).8~Fr.C1.(57~95).9部位经C18反相制备液相色谱柱(25 cm×20 mm, 5 μm)纯化[V(甲醇): V(水)=20: 80]后得化合物2 (19.2 mg).
2.4 糖取代基绝对构型的确定
将5.0 mg化合物1溶于3 mL水中, 加入10 mg蜗牛酶, 37 ℃水浴加热6 h, 水解液用乙酸乙酯萃取(5 mL×3), 水相经冷冻干燥后备用.将5.0 mg化合物2溶于5 mL三氟乙酸溶液中(1 mol/L), 60 ℃水浴加热1 h, 回收溶剂, 残渣经冷冻干燥后备用.将上述样品溶于1 mL吡啶溶液, 加入3 mg L-半胱氨酸甲酯盐酸盐, 60 ℃水浴加热1 h.减压回收溶剂后, 加入0.5 mL三甲基硅烷咪唑, 60 ℃继续水浴加热1 h.然后, 加入2 mL水至反应液中, 用正己烷溶液萃取(2 mL×3).有机相浓缩至2 mL, 过滤后进气相色谱分析.称取2 mg单糖标准品(D-葡萄糖, L-鼠李糖), 按照同样方法进行衍生化处理, 过滤后进气相色谱分析.通过比较样品与对照品的保留时间(D-葡萄糖和L-鼠李糖的保留时间分别为20.56和16.61 min), 确定单糖的绝对构型.
2.5 对α-葡萄糖苷酶的抑制作用
测试样品为已配制好的10 µmol•L-1的化合物储备液.在实验组和空白组分别加入0.1 mol•L-1 PBS缓冲液50和80 μL; 各加入样品10 μL, α-葡萄糖苷酶20 μL, 37 ℃恒温摇床中反应5 min, 再各加入4-硝基苯基-α-D-吡喃葡萄糖苷(pNPG, 1 mmol•L-1) 20 μL, 然后37 ℃恒温摇床中反应15 min, 最后用0.4 mol•L-1 Na2CO3 50 μL终止反应.酶标仪400 nm测OD值.计算样品对α-葡萄糖苷酶的抑制率:
抑制率(%)=(OD空白组-OD给药组)/OD空白组×100%.
2.6 对蛋白酪氨酸磷酸酶PTP1B的抑制作用
测试样品为已配制好的10 µmol•L-1的化合物储备液.采用对硝基苯酚磷酸酯(pNPP)作为人基因重组PTP1B的底物.将化合物与酶在室温下孵育5 min, PTP1B催化的pNPP水解反应在100 μL反应体系中测定.每个反应液含有50 mmol•L-1 4-羟乙基哌嗪乙磺酸(HEPES), 5 mmol•L-1二硫苏糖醇(DTT), 150 mmol•L-1 NaCl, 2 mmol•L-1乙二胺四乙酸(EDTA)以及2 mmol• L-1 pNPP, pH为7.0;在30 ℃下孵育10 min; 加入50 μL 3 mol•L-1的氢氧化钠溶液终止酶反应.水解产物对硝基苯酚钠在405 nm处有很强的光吸收, 采用酶标仪测出各孔的OD值, 即可计算出测试样品对酶促水解的抑制作用.
辅助材料(Supporting Information) 化合物1, 1a和2的UV, IR, 1H NMR, 13C NMR, HR-ESIMS, HSQC, HMBC, 1H-1HCOSY和ECD谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
中国科学院中国植物志编辑委员, 中国植物志, 第78卷, 科学出版社, 北京, 1987, pp. 25~27.Chinese Flora Editorial Board of Chinese Academy of Sciences Flora of China, Vol. 78, Science Press, Beijing, 1985, pp. 25~27(in Chinese).
-
[2]
国家药典委员会, 中国药典(第一部), 中国医药科技出版社, 北京, 2015, p. 161, p. 162.Chinese Pharmacopoeia Commission Chinese Pharmacopoeia, Vol. 1, China Medical Science and Technology Press, Beijing, 2015, p. 161, p. 162(in Chinese).
-
[3]
Koonrungsesomboon, N.; Na-Bangchang, K.; Karbwang, J. Asian Pac. J. Trop. Med. 2014, 7, 421. doi: 10.1016/S1995-7645(14)60069-9
-
[4]
Yu, Y.; Jia, T. Z.; Cai, Q.; Jiang, N.; Ma, M. Y.; Min, D. Y.; Yu, Y. J. Ethnopharmacol. 2015, 160, 211. doi: 10.1016/j.jep.2014.10.066
-
[5]
Kitajima, J.; Kamoshita, A.; Ishikawa, T.; Takano, A.; Fukuda, T.; Isoda, S.; Ida, Y. Chem. Pharm. Bull. 2003, 51, 673. doi: 10.1248/cpb.51.673
-
[6]
Duan, J. A.; Wang, L. Y.; Qian, S. H.; Su, S. L.; Tang, Y. P. Arch. Pharm. Res. 2008, 31, 965. doi: 10.1007/s12272-001-1252-z
-
[7]
Tanaka, K.; Ina, A. Nat. Prod. Commun. 2009, 4, 1095.
-
[8]
Chen, Y. J.; Wu, Y. X.; Wang, H. X.; Gao, K. Fitoterapia 2012, 83, 199. doi: 10.1016/j.fitote.2011.10.015
-
[9]
Kamauchi, H.; Kinoshita, K.; Takatori, K.; Sugita, T.; Takahashi, K.; Koyama, K. Tetrahedron 2015, 71, 1909. doi: 10.1016/j.tet.2015.02.041
-
[10]
Xu, K.; Jiang, J. S.; Feng, Z. M.; Yang, Y. N.; Li, L.; Zang, C. X.; Zhang, P. C. J. Nat. Prod. 2016, 79, 1567. doi: 10.1021/acs.jnatprod.6b00066
-
[11]
He, J.; Shen, Y.; Jiang, J. S.; Yang, Y. N.; Feng, Z. M.; Zhang, P. C.; Yuan, S. P.; Hou, Q. Carbohydr. Res. 2011, 346, 1903. doi: 10.1016/j.carres.2011.06.015
-
[12]
He, J. Y.; Zhu, S.; Komatsu, K. Phytochem. Anal. 2014, 25, 213. doi: 10.1002/pca.v25.3
-
[13]
Hu, Y. C.; Wang, K. Z.; MacMillan, J. B. Org. Lett. 2013, 15, 390. doi: 10.1021/ol303376c
-
[14]
Xu, K.; Yang, P. F.; Yang, Y. N.; Feng, Z. M.; Jiang, J. S.; Zhang, P. C. Org. Lett. 2017, 19, 686. doi: 10.1021/acs.orglett.6b03855
-
[15]
冯卫生, 王建超, 何玉环, 郑晓珂, 宋楷, 张艳丽, 李孟, 赵威, 中国药学杂志, 2015, 50, 2103. http://d.wanfangdata.com.cn/NSTLQK/NSTL_QKJJ0231481181/Feng, W. S.; Wang, J. C.; He, Y. H.; Zheng, X. K.; Song, K.; Zhang, Y. L.; Li, M.; Zhao, W. Chin. Pharm. J. 2015, 50, 2103(in Chinese). http://d.wanfangdata.com.cn/NSTLQK/NSTL_QKJJ0231481181/
-
[1]
-

图 2 化合物1和2的HMBC和1H-1HCOSY二维谱的关键相关信号
Figure 2 Key HMBC and 1H-1HCOSY correlations of 1 and 2

图 3 化合物1a的纽曼投影式和1D NOESY相关
Figure 3 Newman projections and 1D NOESY correlations of compound 1a
表 1 化合物1和2的1H和13C NMR数据(DMSO-d6)
Table 1. 1H NMR spectroscopic data for compounds 1 and 2 in DMSO-d6
No. 1a 1ab No. 2a δH (J in Hz) δC δH (J in Hz) δC δH (J in Hz) δC 1 4.06, brs 60.8 4.11, dd (2.0, 4.5) 62.7 2 160.5 2 6.49, dt (16.0, 4.5) 149.4 6.35, dt (16.0, 4.5) 147.7 3 6.26, d (9.5) 113.3 3 5.86, d (16.0) 106.2 5.81, brd (16.0) 108.8 4 7.86, d (9.5) 144.4 4 80.6 80.4 5 7.27, s 113.0 5 73.5 74.8 6 143.2 6 74.0 74.7 7 152.9 7 80.6 80.6 8 7.10, s 100.4 8 6.20, d (16.0) 109.3 5.88, d (16.0) 109.9 9 150.1 9 6.38, dd (16.0, 5.0) 147.2 6.40, dd (16.0, 5.0) 149.4 10 111.2 10 4.52, dd (5.0, 2.5) 77.3 4.41, dt (5.0, 2.5) 72.2 11 3.87, s 56.3 11 3.29~3.34, m 73.1 3.42, dd (8.0, 2.5) 74.6 1′ 4.91, d (7.5) 100.4 12 3.51~3.61, m 70.8 3.61~3.66, m 72.9 2′ 3.26~3.29, m 73.1 13a 3.51~3.61, m 63.1 3.57, dd (11.5, 5.0) 64.9 3′ 3.23~3.26, m 76.8 13b 3.35~3.43, m 3.74, dd (11.5, 5.0) 4′ 3.06~3.12, m 69.9 1′ 4.08, d (7.5) 101.8 5′ 3.48~3.51, m 75.6 2′ 3.02~3.07, m 73.5 6′a 3.84, d (11.5) 66.4 3′ 3.09~3.12, m 76.7 6′b 3.37~3.40, m 4′ 2.95~3.01, m 70.6 1'' 4.51, d (1.0) 100.5 5′ 3.13~3.17, m 76.6 2'' 3.50~3.53, m 70.4 6′a 3.69, brd (11.5) 61.5 3'' 3.45, dd (9.0, 3.0) 70.7 6′b 3.35~3.43, m 4'' 3.13~3.19, m 72.0 5'' 3.39~3.42, m 68.4 6'' 1.08, d (6.0) 17.9 a在DMSO-d6溶液中测得的数据; b在MeOH-d4甲醇溶液中测得的数据.  下载: 导出CSV
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 3
- 文章访问数: 2083
- HTML全文浏览量: 184
 下载:
下载:
