 图 1
α-手性胺合成策略
Figure 1.
Synthetic strategies for α-chiral amine
图 1
α-手性胺合成策略
Figure 1.
Synthetic strategies for α-chiral amine
引用本文:
陈雕, 徐明华. 过渡金属催化的有机硼试剂对亚胺的不对称加成研究进展[J]. 有机化学,
2017, 37(7): 1589-1612.
doi:
10.6023/cjoc201704017

Citation: Chen Diao, Xu Ming-Hua. Transition Metal-Catalyzed Asymmetric Addition of Organoboron Reagents to Imines[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1589-1612. doi: 10.6023/cjoc201704017
Citation: Chen Diao, Xu Ming-Hua. Transition Metal-Catalyzed Asymmetric Addition of Organoboron Reagents to Imines[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1589-1612. doi: 10.6023/cjoc201704017
过渡金属催化的有机硼试剂对亚胺的不对称加成研究进展
English
Transition Metal-Catalyzed Asymmetric Addition of Organoboron Reagents to Imines
Abstract:
Chiral amines are important building blocks in organic synthesis, they also present in numerous natural products, biologically active compounds and pharmaceutical agents. In recent years, the use of various organoboron reagents in transition metal-catalyzed reactions has attracted considerable attentions because of their ready avilibility, low toxicity, good air and moisture stability as well as high functional group compatibility. This review summarizes the remarkable progress and advances in transition metal-catalyzed asymmetric addition of organoboron reagents to imines over the past few years, providing an overview of the recent achievements in stereoselective synthesis of α-chiral amines by using chiral auxiliary or chiral catalysis strategies. At the present time, rhodium-based asymmetric catalysis has proven to be the most efficient and reliable approach to furnish highly optically active α-chiral amines. Varieties of chiral ligands including monophosphines, biphosphines, amidomonophosphanes, phosphoramidites, phosphites, dienes, sulfur-olefins, phosphorus-olefins have showed the great potential in many asymmetric additions of organoboron reagents to imines, enabling the access of both secondary and tertiary α-chiral amines with good to excellent enantioselectivities. On the other hand, important progress has been made in developing effective palladium catalysts mainly based on chiral pyridine-oxazoline and phosphine-oxazoline ligands. However, future studies in this area towards the objective of developing more efficient, practical and general methods remain challenging.
-
Key words:
- asymmetric synthesis
- / chiral amine
- / metal catalysis
- / asymmetric addition
- / organoboron
-
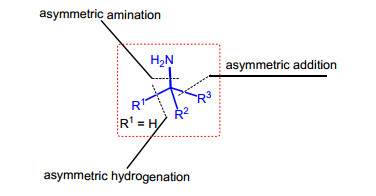
手性胺结构广泛存在于各种天然产物、生物活性分子及药物分子中[1], 因此发展高效的构建手性胺类化合物的方法, 一直都是合成有机化学家们关注的重点.近几十年来, 随着手性技术的发展, 利用不对称反应实现手性胺化合物的高立体选择性合成取得了重要进展.对于α-手性胺类化合物的制备, 依据合成策略, 主要可以概括为以下三类: (1) 不对称氨基化[2]; (2) 前手性烯胺、亚胺的不对称还原(氢化)[3]; (3) 有机富电子试剂[4a, 4b]或者有机金属试剂[4c~4f]对亚胺的不对称加成(图 1).其中, 通过不对称还原或氢化反应策略只能得到含二取代和三取代碳手性中心的α-手性胺, 而不对称氨基化和不对称加成策略则可以合成含季碳手性中心的手性胺类化合物.
图 1
α-手性胺合成策略
Figure 1.
Synthetic strategies for α-chiral amine
虽然利用有机金属试剂对亚胺进行不对称加成是获得高光学纯度手性胺的有效方法, 但传统的有机金属试剂大都存在一些固有的缺点或不足, 例如价格昂贵、毒性较大、对水和空气敏感, 甚至需要现场制备, 不利操作等.近年来, 有机硼试剂由于低毒、稳定、可以直接从市场购买等优点, 在有机合成中的应用越来越受到人们的青睐[5].在合适的过渡金属如铑、钯等催化下, 有机硼试剂通过转金属化生成相应的有机金属活性物种参与反应.本文主要讨论在过去的十来年里, 利用有机硼试剂对亚胺进行不对称加成制备α-手性胺类化合物取得的结果, 分醛亚胺和酮亚胺两个部分展开, 分别描述基于手性辅剂策略和手性金属配合物催化策略的研究进展.
1 醛亚胺的不对称加成
手性二芳基甲胺及手性α-芳基甘氨酸结构是很多生物活性分子及药物分子的重要组成部分, 如:组胺H1受体拮抗剂盐酸西替利嗪, 以及非肽类选择性阿片受体激动剂SNC80具有手性二芳基甲胺的结构骨架[1c]; 抗生素Cefprozil[1d]具有手性α-芳基甘氨酸的结构骨架(图 2).利用过渡金属催化的硼试剂对芳基醛亚胺或者醛酸酯亚胺的不对称加成是合成这两类化合物的一个非常有效的方法.
 图 2
含α-手性胺骨架的生物活性分子
Figure 2.
Bioactive compounds with α-chiral amine motif
图 2
含α-手性胺骨架的生物活性分子
Figure 2.
Bioactive compounds with α-chiral amine motif
1.1 辅剂策略
Ellman小组[6]在2005年报道了铑催化下膦配体促进的芳基硼酸对手性N-叔丁基亚磺酰亚胺的不对称加成反应(Eq. 1).在最初的研究中, 作者发现反应收率与反应物浓度成反比, 而且有较多副产物苯生成, 推测是苯硼酸转金属化后的芳基铑物种直接质子化, 导致亚胺加成反应收率不高.基于这个分析, 通过在6 h内缓慢加入苯硼酸将其浓度维持在一个较低的水平, 果然将反应产率提高到了96%.在底物适应性考查中, 发现无论是富电子还是缺电子硼酸都能取得较好结果, 该反应体系还适用于脂肪亚胺的加成.此外, 该课题组还进一步实现了以醛为起始原料先形成亚胺再进行不对称芳基化的一锅煮反应, 同样取得了很好的收率以及立体选择性.
同年, Batey等[7]也利用类似策略, 发展了一套新的反应体系用于芳基硼酸对手性N-叔丁亚磺酰醛亚胺的不对称加成反应.相比Ellman小组的反应体系, 不需要外加膦配体, 也无需缓慢加入硼酸, 反应在室温下即可进行并能取得较好的收率和同样优秀的非对映选择性(Eq. 2).
2006年, Ellman小组[8]又报道了一价铑催化下芳基硼酸对乙醛酸酯亚胺的不对称加成反应, 用于手性α-芳基甘氨酸的合成, 取得了61%~90%收率, 96%~98% de.研究发现, 硼酸芳环上取代基的电性变化对反应收率及立体选择性的影响不大.所得产物在不同的反应条件下可以选择性地脱除亚磺酰基或者水解酯基, 在NaBH4的条件下经还原得到手性β-氨基醇, 同时可以方便地与亮氨酸甲酯发生缩合反应得到二肽(Eq. 3, Scheme 1).
 图式1
芳基甘氨酸酯产物的衍生化
图式1.
Derivation of the arylglycine ester product
图式1
芳基甘氨酸酯产物的衍生化
图式1.
Derivation of the arylglycine ester product
基于同样的辅剂诱导策略, Ellman等[9]随后又实现了铑催化下烯基氟硼酸钾对芳香醛亚胺及脂肪醛亚胺的不对称加成, 合成了一系列高光学纯度的α-取代烯丙胺.反应的底物适应性较广, 各种取代的烯基氟硼酸钾均能取得很好结果.当反应规模扩大至10 mmol时, 仍然能取得同样的收率及de值(Eq. 4).
由于烯基氟硼酸钾在反应的过程中会涉及到自身脱硼、氧化及多聚化等副反应, 因此找到另外一种更加稳定的硼试剂可能会提高反应收率.确实, 当用BMIDA硼酸酯作为硼试剂时, 相应的不对称烯基化反应可以取得最高98%收率和98% de[10].相比较烯基氟硼酸钾的结果, 利用BMIDA硼酯试剂的每个实施例的收率及de值都更好; 对于收率不理想的多取代的烯基硼试剂及缺电子芳基烯基硼试剂参与的反应, 尤其可以取得较高收率(Eq. 5).
以该不对称烯基化反应为关键步骤, Ellman小组[11]通过6步27%总收率成功实现了天然产物(-)-Aurantio-clavine的高效不对称合成, 而此前文献报道需要13步反应且总收率<1% (Scheme 2).
 图式2
(-)-Aurantioclavine的高效不对称合成
图式2.
Efficient asymmetric synthesis of (-)-aurantiocla-vine
图式2
(-)-Aurantioclavine的高效不对称合成
图式2.
Efficient asymmetric synthesis of (-)-aurantiocla-vine
不同于Ellman等[8]的铑催化方法, Lu小组[12]在2007年报道了阳离子型钯物种催化下的芳基硼酸对乙醛酸酯亚胺的不对称加成反应, 也成功实现了手性α-芳基甘氨酸的不对称合成.芳基钯物种相对铑物种来说亲核性更弱, 所以其大多用于碳-碳键及碳-杂键生成的偶联反应中.相对于中性钯物种, 阳离子钯中心有空置的配位点, 同时具有更“硬”的配位性质, 所以其在反应中可以起到路易斯酸的作用从而活化亚胺以提高反应收率(Eq. 6).
1.2 手性配体策略
之前所述的辅剂策略其优势在于产物的叔丁亚磺酰基能在温和的反应条件下方便地脱除[6, 8], 但缺点在于需要当量的手性源, 不够经济.因此通过合理设计手性配体, 利用催化量的手性催化剂实现反应的对映选择性控制, 相比辅剂策略而言原子经济性更好, 也是当前合成化学发展的必然趋势.
1.2.1 手性膦-酰胺配体
2004年, Tomioka小组[13]设计了一类基于L-缬氨酸骨架的膦-酰氨配体(L1a), 用于一价铑催化的芳基硼酐对N-Ts醛亚胺的不对称加成反应.这是最早的铑催化的硼试剂对亚胺加成的成功例子.有意思的是, 反应适用于位阻较大的芳香亚胺, 芳基邻位取代的亚胺及1-萘亚胺可以取得最好的对映选择性, 而其它底物则结果并不理想(Eq. 7).
在进一步的研究中, 该小组[14]发现增大配体中与磷原子相连芳基的位阻有利于反应的立体选择性控制, 使用配体L1b可以明显提高产物的ee值, 但由于催化活性有一定程度的降低, 需要在反应体系中加入4 Å分子筛来抑制反应时间延长导致的亚胺水解.与之前的结果相比, 这类改进的配体对底物的适应性较好, 除邻位有位阻的芳基亚胺大都能取得较好的结果(Eq. 8).膦酰基保护基也可以在产物ee值保持的情况下温和地脱除.
在对反应过渡态的思考方面[15], 作者认为转金属化后的芳基优先处于磷原子的反位, 这样亚胺从酰基氧的反位与铑配位, TS-1中配体分子中磷原子上的苯基与亚胺氮上的芳基膦酰基存在明显的位阻, 所以TS-2所示为更有利的配位模式(Eq. 9).这也解释了增加配体中磷原子上取代基的位阻能提高反应的立体选择性.
之后, Tomioka小组[16]对该反应又进行了进一步的条件优化, 发现选用三苯基硼烷作为亲核试剂, 可以将反应时间缩短至最快1 h, 从而有效地避免了亚胺水解.反应普遍能取得良好的收率(84%~92%)和对映选择性(90%~96% ee) (Eq. 10).
1.2.2 手性膦配体
2005年, Ellman小组[6]利用(R, R)-DeguPHOS作为手性配体, 实现了铑催化的芳基硼酸对苯甲醛膦酰亚胺的不对称加成反应, 取得了87%~97%的收率和88%~94% ee (Eq. 11).
相比芳基亚胺, 脂肪亚胺由于很容易在酸性或碱性条件下互变为烯胺以及发生自身缩合反应, 所以其不对称芳基化反应往往更加挑战.基于上述反应的催化体系, Ellman小组[17]经过细致的研究, 成功实现了铑催化下DeguPhos促进的芳基硼酸对脂肪醛亚胺的不对称加成反应.他们发现将配体与铑的比例提高到1.4:1, 可以有效地提高反应的收率及立体选择性, 而且在原位形成催化剂后加入硼酸, 保持90 min后再加入亚胺就可以有效地避免亚胺自身缩合.反应的底物适应性较好, 各种类型的脂肪亚胺都能在反应中给出良好的收率和对映选择性(Eq. 12).
2006年, Feringa, Minnaard等[18]报道了N, N-二甲基氨磺酰亚胺的不对称芳基化反应.在最初的研究中, 发现简单的MonoPhos与铑的配合物就能催化反应并取得82% ee, 结合组合化学策略对配体进行大量筛选, 发现对-甲氧基苯胺衍生的亚磷酰胺L3是最好的配体.在最优条件下, 硼酸用量可以低至1.3 equiv., 反应能取得72%~98%收率及82%~95%的对映选择性.不足的是, 反应对亚胺底物及硼酸的电子性质和空间位阻相对敏感, 有一定的局限性.相比Ts基团, 产物胺基上的N, N-二甲基氨磺酰基可以在微波反应条件下更方便地脱除(Eq. 13).
同年, Zhou等[19]发现单亚磷酸酯类配体可以促进一价铑催化的芳基硼酸对苯甲醛亚胺的不对称加成反应(Eq. 14).研究表明, 铑源的不同对反应结果影响很大, 以[Rh]/L4配合物为催化剂, KF为添加剂时反应可以取得较高收率(56%~85%)和对映选择性(85%~96% ee).作者认为配体中手性螺环骨架所提供的较大的二面角对反应的立体选择性起着决定性作用.
2009年, Yamamoto和Miyaura等[20]设计了双齿亚膦酰胺配体L5, 研究了其在铑催化的芳基硼酸对N-Ts及N-Ns苯甲醛亚胺的不对称加成中的应用, 反应可以取得61%~99%的收率及18%~99%的对映选择性.作者认为反应可能存在一定的位阻需求, 因为两个反应底物中如果其中一个为不带任何取代基的苯环时通常只能取得低于90% ee (Eq. 15).
2016年, Kim等[21]将其发展的双环桥头亚膦酰胺类配体L6成功应用于铑催化的芳基硼酸对芳基醛亚胺的不对称加成反应中, 取得了65%~98%收率以及87%~96% ee (Eq. 16).从底物适应性来看, 亚胺原料芳环上的取代基电性及位阻效应对反应影响不大; 但当用带有吸电子及大位阻取代基的芳基硼酸时反应不能发生, 没有得到加成产物.
1.2.3 手性双烯配体
1.2.3.1 不对称芳基化反应
2004年, Hayashi小组[22]首次将手性双烯作为配体用于铑催化的芳基硼酐对N-Ts保护的苯甲醛亚胺的不对称加成反应中, 利用C2对称双环[2.2.2]-辛二烯L7a, 反应可以取得非常出色的结果(最高98%收率, 99%ee), 而且底物适应性也很好, 亚胺以及硼酐芳环上取代基的空间和电子效应对反应的产率及选择性都没有明显影响(Eq. 17).与其它类型的配体相比, 手性双烯在这类反应中表现出更加优秀的催化活性和对映选择性.由于氮上Ts保护基通常难以脱除, 他们又发展了另外一类双环[3.3.1]-壬二烯骨架的手性双烯配体L8, 实现了对N-Ns (Ns=4-硝基苯磺酰基)亚胺的不对称芳基化, 取得了最高96%收率, 99% ee [23](Eq. 18).
对于反应产物的立体化学, 如Eq. 19所示, 推测转金属化后得到的芳基铑物种与亚胺底物可能有两种配位方式, 由于过渡态TS-1中亚胺氮上的磺酰基与配体双键上的苯基有较大的空间位阻, 所以TS-2为优势过渡态, 芳基迁移后得到主要为S构型的产物[22].
基于双环[2.2.2]-己二烯骨架, Hayashi小组[24]之后又成功发展了一类C1对称的手性双烯配体L9a.该配体在芳基硼酐对芳基醛亚胺的不对称加成反应中同样显示出了非常好的催化活性, 催化剂用量甚至可以低至0.3 mol%, 反应收率和对映选择性均极其优异(90%~98%收率, 97%~99.5% ee) (Eq. 20).该配体具有高催化活性的原因可能在于引入吸电子的酯基后, 双键更加缺电子, 有利于转金属化, 由于转金属化是反应的决速步, 所以该配体表现出更好的催化活性.
利用双烯配体L9b, 在相同的反应体系下, Woodward等[25]实现了磺酰二亚胺底物的双不对称芳基化, 以较高的立体选择性得到相应的手性磺酰二胺化合物.这些产物在吡啶/水体系回流条件下可以脱去二氧化硫得到手性二芳基甲胺, 但反应有不同程度的消旋化发生(Eq. 21).
为避免上述保护基脱除过程中产物的部分消旋化, 该小组[26]又设计了在亚胺氮上引入另外一种能在温和条件下方便地脱除的N-叔丁基磺酰胺基(Eq. 22).但在配体L9b促进的芳基化反应条件下, 部分底物由于亚胺水解导致收率大大降低, 有意思的是, 当换用C2对称的手性双烯L7a为配体时, 反应可以取得较高的收率.
基于C1对称的手性双烯配体L9b表现出来的高催化活性, Morimoto等[27]设计了一个串联的不对称芳基化-氨羰基化环合反应用于手性异吲哚啉类化合物的合成.该方法避免了一氧化碳气体的使用, 利用芳基醛或者多聚甲醛为羰基源, 反应中间体无需分离, 底物适应性好, 产物的收率和光学纯度均不错(Eq. 23).
2007年, Lin和Xu等[28]报道了一类结构简单的基于双环[3.3.0]结构的全新手性双烯配体L10, 分子中两个顺并的环戊烯环在空间上呈楔形, 在催化过程中, 两个烯烃双键与金属铑配位可以提供一个对反应的选择性控制非常有利的手性环境, 尤其在铑催化的芳香醛亚胺的不对称芳基加成中能取得极其优秀的对映选择性, 几乎所有产物的光学纯度都在98%~99% ee (Eq. 24).
带邻位酯基的苯甲醛亚胺同样能很高效地发生反应, 在相同的反应条件下经不对称芳基加成后原位内酰氨化, 可以直接合成高光学纯度的手性异吲哚啉化合物(Eq. 25), 而这类化合物中3-位手性的构建用其它催化方法通常是比较困难的.
进一步的研究发现, 利用硼酸/KHF2/H2O的组合反应体系[29], 在手性双环[3.3.0]-辛二烯配体L10促进下, 可以实现对N-Ns亚胺的高对映选择性芳基加成, 以最高96%收率和99% ee得到相应的二芳基甲胺产物[30] (Eq. 26).
随后, 该催化体系又被成功用于芳基硼酸对吲哚醛亚胺的不对称加成反应中, 产物的光学纯度最高可以达到99% ee[31] (Eqs. 27, 28).值得一提的是, 反应能很好地适用于各种缺电性底物, 为合成传统的Friedel-Crafts策略难以实现的α-芳基3-吲哚甲胺类化合物提供了一个新的方法.同时, 对于更加挑战的2-吲哚醛亚胺, 该反应体系也能高效地获得目标芳基化产物(72%收率, 98.5% ee).
2010年, Lin, Feng等[32]报道了另外一类C1对称的同时具有桥环和并环结构的手性双烯配体, 发现这类配体也能高效催化芳基硼酸对醛亚胺的不对称加成, 反应几乎都能取得定量的收率, 但立体选择性较之前C2对称的手性双烯配体均有明显的下降(Eq. 29).
脂肪亚胺的不对称芳基加成报道很少, 一般来说, 需要找到一个活性很高的催化剂能促使芳基化反应顺利发生, 同时反应体系中要避免使用酸或碱以减少副反应的发生. 2011年, Lin小组[33]发现利用铑/烯烃配合物[Rh(L10)(OH)]2可以同时满足如上两个条件, 体系中分子筛的加入也能进一步提高收率.反应的底物适应性较好, 并且收率和对映选择性都很出色(87%~99%收率, 91%~99% ee).结合反应后分子内取代环化策略, 该方法可以用于手性2-芳基吡咯烷及2-芳基哌啶类化合物的高效不对称合成(Eqs. 30, 31).此外, 该催化体系同样适用于N-Ns亚胺, 反应都可以取得良好的收率(87%~99%)和极其优秀的对映选择性(>99% ee).
2010年, Du小组[34]报道了一类基于手性联萘骨架的开链的手性双烯配体在铑催化的芳基醛亚胺的不对称芳基加成反应中的应用.研究发现, 配体分子中联萘骨架上3, 3'位的取代对催化剂的活性和立体选择性有明显的影响, 当3, 3'位无取代时, 该类双烯配体几乎不能催化反应.从反应的底物适应性来看, 该催化体系似乎仅适用于给电性很强的对位烷氧基取代的苯硼酸; 当选用对甲基苯硼酸时, 收率下降至48% (Eq. 32).
2014年, Wu小组[35]报道了另一类C1对称手性双烯配体L13a促进的铑催化的芳基硼酸对芳香醛亚胺的不对称加成反应, 该反应同时适用于N-Ts及N-Ns亚胺并且底物适应性较好; 催化剂具有较高的反应活性, 反应1 h便可完全, 可以以56%~99%收率, 90%~99% ee得到手性二芳基甲胺产物.在亚胺底物中引入合适的官能团, 其加成产物经过几步简单转化可用于手性四氢异喹啉类化合物的合成(Eq. 33, Scheme 3).
 图式3
手性四氢异喹啉类化合物的合成
图式3.
Synthesis of tetrahydroisoquinoline
图式3
手性四氢异喹啉类化合物的合成
图式3.
Synthesis of tetrahydroisoquinoline
1.2.3.2 不对称烯基化及烯丙基化反应
手性双烯配体不仅适用于醛亚胺的不对称芳基化反应, 同时还能用于高效的不对称烯基化以及烯丙基化反应. 2012年, Lam小组[36]报道了铑催化下手性双烯配体促进的烯基氟硼酸钾对醛亚胺的不对称加成反应(Eq. 34).在最初的研究中, 以苯甲醛亚胺为底物, 虽然反应可以顺利发生, 但产物的ee值都不理想, 最好仅在55%.考虑到在烯基化反应过程中亚胺C=N双键Z/E构型变化的可能影响, 作者尝试了以C=N双键构型固定的环状磺酰醛亚胺为反应底物进行不对称烯基化(Eq. 35).果然, 反应结果有了明显的提高, 不同的烯基氟硼酸钾都能实现对各种环状醛亚胺的不对称加成, 并且给出中等到优秀的收率(50%~94%)以及出色的对映选择性(94%~99% ee).值得一提的是, 在相同的反应体系下, 五元环状磺酰酮亚胺的不对称烯基化反应也能取得68%收率和90% ee.
上述催化体系也可以进一步用于不对称烯丙基化反应[37].利用烯丙基氟硼酸钾或者取代的烯丙基氟硼酸钾对环状磺酰醛亚胺及酮亚胺进行不对称加成, 反应能取得最高97%收率, 98%ee (Eq. 36).将烯丙基氟硼酸钾换成烯丙基硼酸频那醇酯, 发现反应只能取得28% ee, 证明这里氟硼酸钾试剂非常关键, 但具体原因并不清楚.
深入研究发现, E-crotyltrifluoroborate与亚胺加成后主要得到反式产物, 而Z-crotyltrifluoroborate与亚胺加成之后则得到顺式为主的产物.由此, 作者推测反应经历了六元环状过渡态.进一步的氘代实验结果表明反应中烯丙基铑物种在快速互变(Scheme 4).
 图式4
氘代实验
图式4.
Deuterium labeling experiment
图式4
氘代实验
图式4.
Deuterium labeling experiment
2014年, Lin, Feng等[38]以铑/双环[3.3.0]手性双烯络合物为催化剂, 成功实现了烯基氟硼酸钾对非环状的苯甲醛亚胺的高立体选择性加成, 得到一系列近乎光学纯的手性烯丙胺(97%~99% ee) (Eq. 37).反应具有很好的底物适应性, 对于各种取代的烯基氟硼酸钾、环已烯氟硼酸钾及苯乙烯基氟硼酸钾均能取得优异的结果, 芳环上的取代基的空间位阻和电子性质对反应都没有明显影响.
几乎同时, Wu小组[39]也报道了双烯配体L13b促进的烯基氟硼酸钾对苯甲醛亚胺的不对称加成反应, 底物适应性也比较广, 取得了73%~97%的收率和72%~99% ee.加成产物还原后经分子内Mitsonubu反应可以用于邻位芳基取代的吡咯烷类化合物的合成(Scheme 5).
 图式5
手性烯丙基胺的合成与衍生
图式5.
Synthesis and derivation of chiral allylic amines
图式5
手性烯丙基胺的合成与衍生
图式5.
Synthesis and derivation of chiral allylic amines
1.2.4 杂原子-烯烃配体
2011年, Shintani、Hayashi等[40]设计了一类五元环状膦烯配体, 在铑催化的芳基硼酸对苯甲醛亚胺的不对称加成反应中, 取得了不错的结果, 尤其是反应的底物适应性很好, 不仅适用于芳基和杂芳基的亚胺底物, 脂肪亚胺也能取得较好的结果(Eq. 38).
2013年, Xu小组[41]以简单手性硫烯为配体, 成功实现了铑催化下芳基硼酸对六元环状磺酰醛亚胺的不对称加成反应.在室温条件下, 反应即可高效高选择性地进行, 以优秀的收率(93%~99%)得到一系列近乎光学纯的芳基化产物(97%~99% ee).对反应过渡态的分析认为与双键相连的R基团的空间位阻作用使得环状醛亚胺的环完全倾向于处在远离R的位置(B中所示), 这样, 芳基从亚胺re-面迁移, 从而得到(R)构型的产物(Eq. 39, 图 3).
 图 3
推测的反应过渡态
Figure 3.
Proposed reaction transition state
图 3
推测的反应过渡态
Figure 3.
Proposed reaction transition state
最近, Xu等[42]又成功实现了铑催化下硫烯配体L17 促进的芳基硼酐对N, N-二甲基氨磺酰亚胺的不对称加成反应(Eq. 40).这个反应的底物适应性非常好, 克服了之前利用MonoPhos类单膦配体L3[18]、双环桥头亚膦酰胺类配体L6[21]以及开链双烯配体L12[34]的底物局限性, 通过合理的组合, 可以几乎完美地得到各种二芳基甲胺产物, 反应还尤其适用于芳基邻位有取代基的底物, 可以获得97%~99% ee的极高的对映选择性.值得一提的是, 该催化体系也适用于N-Ts亚胺的不对称芳基加成.基于该方法, 两个邻位有羟乙基取代的亚胺经加成反应, 得到光学纯度为99% ee的相应产物, 再经简单转化成功用于治疗尿频的药物分子Solifenacin以及作为多巴胺受体D1探针分子的天然产物(S)-(+)-Cryp-tostyline Ⅱ的高效合成(Eq. 41).该研究也为构建药物研究中重要的手性1-芳基-1, 2, 3, 4-四氢异喹啉骨架提供了一个简单、有效的催化不对称方法.
1.2.5 手性噁唑啉配体
2012年, Zeng小组[43]报道了二价钯催化下双恶唑啉配体促进的芳基硼酸对乙醛酸酯亚胺的不对称加成反应(Eq. 42).有趣的是, 与之前Lu小组报道的需要阳离子型钯催化不同, 反应在Pd(OAc)2/双噁唑啉配体催化下便可以取得较高的收率, 说明噁唑啉类配体不仅可以调控该反应的立体选择性, 同时也可以提高催化剂的反应活性.在10 mol%的催化剂作用下, 该反应对于富电子硼酸能取得较好的结果, 但对于缺电子硼酸反应收率有一定的下降.
2 酮亚胺的不对称加成
酮亚胺经不对称加成反应可以得到含季碳立体中心的α-手性胺类化合物, 但相比前述醛亚胺, 酮亚胺的反应性大大降低, 并且由于反应位点两侧空间区分较小, 所以要实现其立体选择性控制通常更难.
2.1 手性辅剂策略
2011年, Ellman小组[44]报道了铑催化下芳基硼酐对靛红叔丁亚磺酰亚胺的不对称芳基化反应, 以合成含季碳手性中心的3-芳基3-氨基吲哚酮.反应在一价铑催化下顺利进行, 靛红母核上的取代及芳基硼酐上的取代基对反应的影响不大, 以65%~92%收率和中等到好的立体选择性(88%~92% de)得到目标产物(Eq. 43).
2.2 手性配体策略
2.2.1 手性双膦配体
2006年, Shibasaki, Kanai等[45]报道了Cu(Ⅰ)/La(Ⅲ)共催化的环戊基-DuPhos促进的烯丙基频那醇硼酯对酮亚胺的不对称加成反应, 以76%~98%收率, 23%~93% ee得到了一系列α-芳基-高烯丙氨类化合物(Eq. 44).异丙醇锂作为共催化剂可以显著提高反应的活性及立体选择性, 核磁研究表明其可以有效增加活性亲核物种(烯丙基铜试剂)的浓度.苄基保护基可以经IBX氧化成亚胺中间体, 随后在盐酸水溶液中脱除, 产物光学活性保持.
2015年, Jarvo等[46]报道了银催化下Walphos促进的联烯频那醇硼酯对环状磺酰酮亚胺的不对称加成, 成功合成了一系列含季碳手性中心α-芳基/烷基-高炔丙氨类化合物, 取得了90%~98% ee (Eq. 45).
几乎同时, Kong, McLaughlin等[47]报道了一价铑催化的Walphos促进的芳基及烯基硼酸对环状磺酰酮亚胺的不对称加成反应, 芳基酮亚胺及脂肪酮亚胺均能适用于此反应, 取得了40%~99%收率, 88%~99% ee (Eq. 46).
2.2.2 手性烯烃类配体
2010年, Shintani, Hayashi等[48]首次实现了铑催化下双环[2.2.2]手性双烯配体促进的四芳基硼钠试剂对酮亚胺的不对称加成反应, 以53%~97%的收率和94%~99.5% ee得到了一系列含α-季碳手性中心的手性胺.由于酮亚胺的反应性较低, 所以作者采用了亲核能力更强的四芳基硼钠试剂, 反应中铑/双烯络合物催化剂需要事先制备, 体系具有较好的底物适应性(Eq. 47).
在此基础上, 该小组[49]随后又发现可以用更加容易制备的芳基氟硼酸钾试剂代替四芳基硼钠, 并且将亚胺氮上的保护基拓展至易脱除的Ns, 反应同时还适用于杂芳基硼酸及烯基硼酸, 均能取得良好的收率和优异的对映选择性(Eq. 48).
2012年, Nishimura, Hayashi[50]又成功实现了C1-对称的手性双烯配体L9c促进的五元和六元环状磺酰酮亚胺的不对称芳基化反应.同样, 相应的铑/烯烃配合物催化剂需要事先制备, 在其催化下, 可以以70%~93%的收率和93%~99% ee获得一系列含季碳手性中心的苯并磺内酰胺和磺胺内酯类化合物.其中苯并磺胺内酯产物在四氢铝锂的作用下很容易开环得到α-三芳基取代的手性甲胺, 这类化合物由于三个芳基取代基非常相似, 要实现它们的高效不对称合成通常是非常困难的(Eq. 49).
2013年, Xu小组[51]发展了新的支链结构的手性亚磺酰胺-烯烃配体, 成功用于铑催化的硼酸对环状酮亚胺的不对称芳基化反应中, 首次实现了含季碳手性的相应羧基和三氟甲基取代的系列磺内酰胺和磺胺内酯类化合物的高对映选择性合成.利用简单硫烯配体L16, 反应在温和的条件下就可以进行, 产物的对映选择性最高为99% ee (Eq. 50).这里手性硫烯配体相比之前报道的双烯及双膦配体, 结构更加简单, 合成极其方便, 而且反应中无需预先制备好铑配合物.
1, 2, 5-噻二唑啉-1, 1-二氧化物结构广泛存在于生物活性天然产物和药物分子中, 但手性噻二唑啉的合成报道很少.在进一步的研究中, Xu等[52]利用相同的原位生成的手性铑/硫烯配合物为催化剂, 通过环状磺酰二酮亚胺的不对芳基化反应, 成功实现了一系列含四取代手性中心的2, 3-二氢-1, 2, 5-噻二唑啉-1, 1-二氧化物的高对映选择性合成.反应在非常温和的条件下进行, 收率及ee值均高达99% (Eq. 51).产物经一步开环反应可以方便地用于其它方法难以实现的手性α-叔氨酮的构建, 为这类重要化合物的合成提供了新的对映选择性方法.
相比芳香酮亚胺, 烷基取代的脂肪酮亚胺的不对称加成更加挑战.通过对反应条件的优化, Xu小组[53]又利用手性硫烯配体L16成功实现了铑催化下五元和六元环状N-磺酰脂肪酮亚胺的不对称芳基化反应, 成功合成了一系列α-位芳基和烷基取代的苯并磺内酰胺及苯并磺胺内酯.反应具有非常好的底物适用性, 可以取得中等到优秀的收率, 尤其是产物的对映选择性可高达99.9% ee.产物经简单转化, 可以进一步实现含季碳手性中心的四氢异喹啉及苯并恶嗪酮类化合物的高效立体选择性构建, 方法并首次实现了NMDA拮抗剂FR115427的催化不对称合成(Scheme 6).
 图式6
环状N-磺酰基取代烷基酮亚胺的不对称芳基化
图式6.
Asymmetric arylation of cyclic N-sulfonyl aryl alkyl ketimines
图式6
环状N-磺酰基取代烷基酮亚胺的不对称芳基化
图式6.
Asymmetric arylation of cyclic N-sulfonyl aryl alkyl ketimines
随后, 经过反应条件的微调, 在相同的硫烯配体L16促进下, Xu等[54]又实现了苯并噁嗪酮以及喹喔啉酮的不对称芳基化反应, 以极其优异的对映选择性(高达99.9% ee), 成功合成了一系列几乎光学纯的3, 4-二氢-苯并[1, 4]-恶嗪-2-酮以及二氢喹喔啉酮类化合物(Eq. 52).
α, α-二芳基氨基酸是一些具有重要生理活性的天然产物、已上市药物及正在开发的活性化合物的关键核心骨架, 然而其高效的不对称合成方法研究很少有成功的报道. Xu小组[55]设计、发展了一类新型手性膦烯配体, 利用开链结构的L21实现了铑催化下1, 2, 5-噻二唑啉-1, 1-二氧类酮亚胺的不对称芳基化反应, 在温和的条件下, 高效地合成了一系列含季碳手性的偕二芳基取代的磺胺乙内酰脲以及4-乙氧基-2, 3-二氢-1, 2, 5-噻唑-1, 1-二氧类化合物(最高99% ee).该方法操作简单、底物适用范围广、立体选择性高, 产物经简单转化可用于非天然的手性α, α-二芳基氨基酸衍生物的合成以及各种含氮杂环的构建, 并首次实现了Merck公司报道的BACE1抑制剂(R)-iminohydantoin的催化不对称合成(Scheme 7).
 图式7
简单手性膦-烯配体用于环状亚胺的不对称芳基化
图式7.
Simple phosphite-olefin as ligand for asymmetric arylation of cyclic ketimine
图式7
简单手性膦-烯配体用于环状亚胺的不对称芳基化
图式7.
Simple phosphite-olefin as ligand for asymmetric arylation of cyclic ketimine
最近, Xu等[56]又利用不同的烯烃配体, 实现了芳基硼酸对α, β-不饱和环状磺酰酮亚胺的分步1, 4-和1, 2-加成反应, 成功构建了高光学纯度的具有偕二芳基取代的手性磺胺内酯, C1-对称手性双烯配体L7c及手性硫烯配体L16在反应中表现出很高的催化活性和对映选择性.加成产物经简单转化后可以得到两类含季碳手性中心并具有四并环结构的手性胺(Scheme 8).
 图式8
铑催化的α, β-不饱和环状酮亚胺的逐步不对称1, 4-和1, 2-加成
图式8.
Rh-catalyzed stepwise asymmetric 1, 4-and 1, 2-addition of α, β-unsaturated cyclic ketimines
图式8
铑催化的α, β-不饱和环状酮亚胺的逐步不对称1, 4-和1, 2-加成
图式8.
Rh-catalyzed stepwise asymmetric 1, 4-and 1, 2-addition of α, β-unsaturated cyclic ketimines
一般来说, 对于一些稳定性差的亚胺很难进行直接加成反应. 2013年, Hayashi和Nishimura[57]以环状半缩醛胺为起始反应物, 利用硼酐的脱水作用在反应体系中原位形成N-羰基亚胺, 实现了[Rh(OH)(L22a)]2催化下的高对映选择性不对称芳基加成(91%~98% ee), 用于合成各种含有三芳基取代的立体中心的手性异吲哚啉酮化合物(Scheme 9).研究发现, 选用高活性的羟基铑络合物作为催化剂是反应成功的关键, 硼酐在反应中既起到脱水剂的作用, 同时又是芳基加成试剂.
 图式9
3-芳基-3-羟基-1-异吲哚啉酮的不对称芳基化
图式9.
Asymmetric arylation of 3-aryl-3-hydroxyisoindolin-1-ones
图式9
3-芳基-3-羟基-1-异吲哚啉酮的不对称芳基化
图式9.
Asymmetric arylation of 3-aryl-3-hydroxyisoindolin-1-ones
在进一步的研究中[58], 将上述发现的高活性的羟基铑物种[Rh(OH)(L22a)]2用于反应性相对略低的简单环状磺酰亚胺的不对称芳基加成中, 相比原位形成的催化剂, 反应可以取得明显提高的收率(70%~98%)以及优异的对映选择性(91%~99% ee) (Eq. 53).所得产物α-二芳基磺胺内酯经过几步简单转化可以在ee值保持的情况下开环得到各种含季碳手性的胺类合成砌块.
类似的, Lin, Feng等[59]基于双环[3.3.0]手性双烯配体L10, 将活性羟基铑物种[Rh(OH)(L10)]2用于催化环状磺酰脂肪酮亚胺的对映选择性芳基化反应, 实现了一系列含四取代手性中心的磺胺内酯类化合物的高效不对称合成, 最高取得98% ee (Eq. 54).研究发现选择更加稳定的芳基频那醇硼酯作为硼试剂可以相应减少脱硼副反应, 从而提高反应收率, 但当缺电子的芳基硼酯参与反应时, 收率仍有急剧的下降, 且亚胺底物中的烷基取代基的位阻也会对反应收率有明显的影响.同样, 产物在LAH的条件下开环, 可以得到光学活性的β-氨基醇.
2015年, Nishimura等[60]又报道了活性羟基铑物种[Rh(OH)(L22b)]2催化环状磺酰亚胺酯的不对称芳基化反应, 实现了一系列手性偕二芳基取代的苯并磺胺内酯类化合物的高效合成, 取得了83%~97% ee (Eq. 55).产物可以经简单转化, 以中等收率得到ee值保持的α, α-二芳基-α-N-甲基氨基酸酯.
2.2.3 手性含氮配体
2013年, Zhang小组[61]报道了首例二价钯催化下, 吡啶-噁唑啉配体L23促进的环状脂肪酮亚胺的不对称芳基化反应.反应在空气条件下进行, 以利于将生成的Pd0氧化成PdⅡ, 同时选用极性的三氟乙醇为溶剂以帮助加快质子化步骤, 从而提高收率(Eq. 56).反应的底物适应性较好, 吸电子硼酸也能取得较高的收率, 但芳基邻位有取代的硼酸对反应收率和选择性有明显的影响, 当选用2-甲氧基苯硼酸时反应在48 h后也仅有约15%的核磁收率.
反应的立体选择性可以通过如下模型来解释(Eq. 57), 转金属化后的钯物种中, 芳基处于噁唑啉环的对位, 而亚胺则从吡啶环的对位与铑配位, 为避免环上SO2基团与配体中的叔丁基的位阻, 主要采取过渡态Ⅱ中的取向, Ar从亚胺的Re面进攻, 得到相应构型的产物.
2014年, Lu, Hayashi等[62]也报道了钯催化的环状酮亚胺的不对称芳基化.相比五元环系底物, 六元环状亚胺的反应性通常要低, 但加成产物却更容易经开环得到各种衍生物.对多种手性配体包括膦-噁唑啉、吡啶-噁唑啉、双膦、双烯等的考察结果表明, 手性膦-噁唑啉配体i-Pr-Phox可以给出最好的反应结果.值得一提的是, 反应条件中添加物AgBF4, AgSbF6, K3PO4及质子海绵的选择对收率和选择性非常关键, 一些情况下产物会发生消旋化, 因此还需要AgBF4或AgSbF6与碱性的K3PO4或质子海绵的组合以中和反应体系中的酸性.在各自的最优反应条件下, 反应大都可以取得很高的收率和极其优秀的对映选择性(96%~99.9% ee) (Eq. 58).
2015年, Zhang, Xie等[63]又报道了二价钯催化下吡啶-恶唑啉配体L23促进的环状磺酰亚胺酯的不对称芳基化反应.催化体系能实现优异的立体选择性控制(95%~99% ee), 但对于大位阻芳基硼酸、杂芳基硼酸及带有吸电子基团的芳基硼酸反应收率会有不同程度的下降. DFT计算表明反应中芳基插入亚胺的一步是反应的决速步, 也是选择性控制的关键步(Eq. 59).
在一系列钯催化的不对称芳基加成研究取得突破之后, Lassaletta, Fernández, Monge等[64]进一步设计了一类新型的手性吡啶-腙配体, 也成功应用于之前报道的各种环状磺酰酮亚胺, 包括芳香亚胺、脂肪亚胺、酯基取代亚胺及磺酰二酮亚胺等的不对称芳基化反应中, 取得了优良的收率和对映选择性(Eqs. 60, 61).
虽然过渡金属催化的酮亚胺的不对称加成近年来取得了很大的进展, 但是吲哚酮亚胺却一直是一类非常挑战的底物, 利用芳基硼试剂对其进行加成此前并未有成功的报道.最近, Zhang小组[65]通过对底物中吲哚和亚胺氮上的保护基的详细考察以及系列手性吡啶-恶唑啉配体的筛选, 实现了钯催化下芳基硼酸对该类底物的不对称加成反应.研究发现, In-Pyrox为最优配体, 当底物分子中吲哚和亚胺氮上分别为2, 6-二氯苄基和叔丁基磺酰基时, 在三氟乙醇溶剂中, 70 ℃条件下, 反应可以取得了90%~98% ee (Eq. 62).
3 总结与展望
由于α-手性胺在有机合成及药物研究中的重要用途, 化学家们对它们的不对称合成方法的探索乐此不疲.综上所述, 基于手性辅剂及手性配体策略的过渡金属催化的有机硼试剂对亚胺的不对称加成研究在近十多年里已经取得了极大的进展, 尤其是化学家们成功发展了一系列高效的手性配体用于反应的立体选择性控制, 其中包括了结构多样的各种膦-氮配体、单膦配体、双膦配体、双烯配体、硫烯配体、磷烯配体、含氮配体等.值得一提的是, 在铑催化的反应中, 手性烯烃类配体展现出尤为独特的催化活性和对映选择性, 而且部分如手性硫烯, 结构极其简单、合成方便, 令人印象深刻, 为一些重要手性胺及相关生理活性分子的合成提供了温和、高效并相对经济的方法.另一方面, 近年来发展起来的基于含氮配体的钯催化反应也非常值得关注, 但目前在研究的深度和广度上还有待于进一步探索.相对于金属铑和钯, 其它廉价过渡金属如铜、镍等催化的有机硼试剂对亚胺的不对称加成研究仍未有突破.在今后的研究中, 设计简单高效的新型手性配体, 开发实用的催化体系, 拓展结构新颖的反应底物, 探索多样性的有机硼试剂, 仍然充满着挑战和机遇.随着人们对相关反应的不对称催化机理的理解和认识的不断深入, 相信这个领域的研究必将取得更大的发展.
-
-
[1]
(a) Calderon, S. N.; Rothman, R. B.; Porreca, F.; Flippen-Anderson, J. L.; McNutt, R. W.; Xu, H.; Smith, L. E.; Bilsky, E. J.; Davis, P.; Rice, K. C. J. Med. Chem. 1994, 37, 2125.
(b) Van Bambeke, F.; Van Laethem, Y.; Courvalin, P.; Tulkens, P. M. Drugs 2004, 64, 913.
(c) Calderon, S. N.; Rice, K. C.; Rothman, R. B.; Porreca, F.; Anderson, J. L. F.; Kayakiri, H.; Xu, H.; Becketts, K.; Smith, L. E.; Bilsky, E. J.; Davis, P.; Horvath, R. J. Med. Chem. 1997, 40, 695.
(d) Wiseman, L. R.; Benfield, P. Drugs 1993, 2, 295. -
[2]
(a) Deng, Q. H.; Xu, H. W.; Yuen, A. W.; Xu, Z. J.; Che, C. M. Org. Lett. 2008, 10, 1529.
(b) Lee, E. C.; Fu, G. C. J. Am. Chem. Soc. 2007, 129, 12066.
(c) Xu, B.; Zhu, S. F.; Zuo, X. D.; Zhang, Z. C.; Zhou, Q. L. Angew. Chem., Int. Ed. 2014, 53, 3913.
(d) Xu, B.; Zhu, S. F.; Xie, X. L.; Shen, J. J.; Zhou, Q. L. Angew. Chem., Int. Ed. 2011, 50, 11483. -
[3]
(a) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029.
(b) Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069.
(c) Wang, D. S.; Chen, Q. A.; Lu, S. M.; Zhou, Y. G. Chem. Rev. 2012, 112, 2557. -
[4]
(a) Wang, J.; Liu, X.; Feng, X. Chem. Rev. 2011, 111, 6947.
(b) Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626.
(c) Marques, C. S.; Burke, A. J. ChemCatChem 2011, 3, 635.
(d) Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600.
(e) Lin, G.-Q.; Xu, M.-H.; Zhong, Y.-W.; Sun, X.-W. Acc. Chem. Res. 2008, 41, 831.
(f) Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984. -
[5]
(a) Suzuki, A. J. Organo. Chem. 1999, 576, 147.
(b) Miyaura, N. Cross-Coupling Reactions. A Practical Guide, Topics in Current Chemistry, Vol. 219, Springer, Berlin, 2002.
(c) Negishi, E. Handbook of Organopalladium Chemistry for Organic Synthesis, Vol. 1, Wiley, New York, 2002.
(d) Liu, Q; Tian, B. ; Tian, P. ; Tong, X. -F. ; Lin, G. -Q. Chin. J. Org. Chem. 2015, 35, 1 (in Chinese).
(刘强, 田兵, 田平, 童晓峰, 林国强, 有机化学, 2015, 35, 1. )
(e) Liu, Y. -Y. ; Zhang, W. -B. Chin. J. Org. Chem. 2016, 36, 2249 (in Chinese).
(刘媛媛, 张万斌, 有机化学, 2016, 36, 2249. ) -
[6]
Weix, D. J.; Shi, Y.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 1092. doi: 10.1021/ja044003d
-
[7]
Bolshan, Y.; Batey, R. A. Org. Lett. 2005, 7, 1481. doi: 10.1021/ol050014f
-
[8]
Beenen, M. A.; Weix, D. J.; Ellman, J. A. J. Am. Chem. Soc. 2006, 128, 6304. doi: 10.1021/ja060529h
-
[9]
Brak, K.; Ellman, J. A. J. Am. Chem. Soc. 2009, 131, 3850. doi: 10.1021/ja9002603
-
[10]
Brak, K.; Ellman, J. A. J. Org. Chem. 2010, 75, 3147. doi: 10.1021/jo100318s
-
[11]
Brak, K.; Ellman, J. A. Org. Lett. 2010, 12, 2004. doi: 10.1021/ol100470g
-
[12]
Dai, H.; Lu, X. Org. Lett. 2007, 9, 3077. doi: 10.1021/ol0711220
-
[13]
Kuriyama, M.; Soeta, T.; Hao, X.; Chen, Q.; Tomioka, K. J. Am. Chem. Soc. 2004, 126, 8128. doi: 10.1021/ja0475398
-
[14]
Hao, X.; Kuriyama, M.; Chen, Q.; Yamamoto, Y.; Yamada, K.; Tomioka, K. Org. Lett. 2009, 11, 4470. doi: 10.1021/ol901866y
-
[15]
Hao, X.; Chen, Q.; Yamada, K.-i.; Yamamoto, Y.; Tomioka, K. Tetrahedron 2011, 67, 6469. doi: 10.1016/j.tet.2011.06.033
-
[16]
Hao, X.; Chen, Q.; Kuriyama, M.; Yamada, K.-I.; Yamamoto, Y.; Tomioka, K. Cat. Sci. Tec. 2011, 1, 62. doi: 10.1039/c0cy00083c
-
[17]
Trincado, M.; Ellman, J. A. Angew. Chem., Int. Ed. 2008, 47, 5623. doi: 10.1002/anie.v47:30
-
[18]
Jagt, R. B.; Toullec, P. Y.; Geerdink, D.; de Vries, J. G.; Feringa, B. L.; Minnaard, A. J. Angew. Chem., Int. Ed. 2006, 45, 2789. doi: 10.1002/(ISSN)1521-3773
-
[19]
Duan, H.-F.; Jia, Y.-X.; Wang, L.-X.; Zhou, Q.-L. Org. Lett. 2006, 8, 2567. doi: 10.1021/ol060755w
-
[20]
Kurihara, K.; Yamamoto, Y.; Miyaura, N. Adv. Synth. Catal. 2009, 351, 260. doi: 10.1002/adsc.200800631
-
[21]
Lee, A.; Kim, H. J. Org. Chem. 2016, 81, 3520. doi: 10.1021/acs.joc.6b00033
-
[22]
Tokunaga, N.; Otomaru, Y.; Okamoto, K.; Ueyama, K.; Shintani, R.; Hayashi, T. J. Am. Chem. Soc. 2004, 126, 13584. doi: 10.1021/ja044790e
-
[23]
Otomaru, Y.; Kina, A.; Shintani, R.; Hayashi, T. Tetrahedron:Asymmetry 2005, 16, 1673. doi: 10.1016/j.tetasy.2005.02.022
-
[24]
Okamoto, K.; Hayashi, T.; Rawal, V. H. Chem. Commun. 2009, 4815.
-
[25]
Crampton, R.; Woodward, S.; Fox, M. Adv. Synth. Catal. 2011, 353, 903. doi: 10.1002/adsc.v353.6
-
[26]
Crampton, R. H.; Fox, M.; Woodward, S. Tetrahedron:Asymmetry 2013, 24, 599. doi: 10.1016/j.tetasy.2013.04.006
-
[27]
Fujioka, M.; Morimoto, T.; Tsumagari, T.; Tanimoto, H.; Nishiyama, Y.; Kakiuchi, K. J. Org. Chem. 2012, 77, 2911. doi: 10.1021/jo300201g
-
[28]
Wang, Z.-Q.; Feng, C.-G.; Xu, M.-H.; Lin, G.-Q. J. Am. Chem. Soc. 2007, 129, 5336. doi: 10.1021/ja0710914
-
[29]
Wang, Z.-Q.; Feng, C.-G.; Zhang, S.-S.; Xu, M.-H.; Lin, G.-Q. Angew. Chem., Int. Ed. 2010, 49, 5780. doi: 10.1002/anie.v49:33
-
[30]
Wang, L.; Wang, Z.-Q.; Xu, M.-H.; Lin, G.-Q. Synthesis 2010, 3263.
-
[31]
Yang, H.-Y.; Xu, M.-H. Chem. Commun. 2010, 46, 9223. doi: 10.1039/c0cc04086j
-
[32]
Shao, C.; Yu, H.-J.; Wu, N.-Y.; Feng, C.-G.; Lin, G.-Q. Org. Lett. 2010, 12, 3820. doi: 10.1021/ol101531r
-
[33]
Cui, Z.; Yu, H.-J.; Yang, R.-F.; Gao, W.-Y.; Feng, C.-G.; Lin, G.-Q. J. Am. Chem. Soc. 2011, 133, 12394. doi: 10.1021/ja2046217
-
[34]
Cao, Z.; Du, H. Org. Lett. 2010, 12, 2602. doi: 10.1021/ol1008087
-
[35]
Chen, C.-C.; Gopula, B.; Syu, J.-F.; Pan, J.-H.; Kuo, T.-S.; Wu, P.-Y.; Henschke, J. P.; Wu, H.-L. J. Org. Chem. 2014, 79, 8077. doi: 10.1021/jo5012653
-
[36]
Luo, Y.; Carnell, A. J.; Lam, H. W. Angew. Chem., Int. Ed. 2012, 51, 6762. doi: 10.1002/anie.201202136
-
[37]
Luo, Y.; Hepburn, H. B.; Chotsaeng, N.; Lam, H. W. Angew. Chem., Int. Ed. 2012, 51, 8309. doi: 10.1002/anie.v51.33
-
[38]
Cui, Z.; Chen, Y.-J.; Gao, W.-Y.; Feng, C.-G.; Lin, G.-Q. Org. Lett. 2014, 16, 1016. doi: 10.1021/ol5000154
-
[39]
Gopula, B.; Chiang, C. W.; Lee, W. Z.; Kuo, T. S.; Wu, P. Y.; Henschke, J. P.; Wu, H. L. Org. Lett. 2014, 16, 632. doi: 10.1021/ol4035897
-
[40]
Shintani, R.; Narui, R.; Tsutsumi, Y.; Hayashi, S.; Hayashi, T. Chem. Commun. 2011, 47, 6123. doi: 10.1039/c1cc11823d
-
[41]
Wang, H.; Xu, M.-H. Synthesis 2013, 45, 2125. doi: 10.1055/s-00000084
-
[42]
Jiang, T.; Chen, W.-W.; Xu, M.-H. Org. Lett. 2017, 19, 2138. doi: 10.1021/acs.orglett.7b00776
-
[43]
Chen, J.; Lu, X.; Lou, W.; Ye, Y.; Jiang, H.; Zeng, W. J. Org. Chem. 2012, 77, 8541. doi: 10.1021/jo301423e
-
[44]
Jung, H. H.; Buesking, A. W.; Ellman, J. A. Org. Lett. 2011, 13, 3912. doi: 10.1021/ol201438k
-
[45]
Wada, R.; Shibuguchi, T.; Makino, S.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 7687. doi: 10.1021/ja061510h
-
[46]
Osborne, C. A.; Endean, T. B. D.; Jarvo, E. R. Org. Lett. 2015, 17, 5340. doi: 10.1021/acs.orglett.5b02692
-
[47]
Kong, J.; McLaughlin, M.; Belyk, K.; Mondschein, R. Org. Lett. 2015, 17, 5520. doi: 10.1021/acs.orglett.5b02032
-
[48]
Shintani, R.; Takeda, M.; Tsuji, T.; Hayashi, T. J. Am. Chem. Soc. 2010, 132, 13168. doi: 10.1021/ja106114q
-
[49]
Shintani, R.; Takeda, M.; Soh, Y. T.; Ito, T.; Hayashi, T. Org. Lett. 2011, 13, 2977. doi: 10.1021/ol200958q
-
[50]
Nishimura, T.; Noishiki, A.; Tsui, G. C.; Hayashi, T. J. Am. Chem. Soc. 2012, 134, 5056. doi: 10.1021/ja300697c
-
[51]
Wang, H.; Jiang, T.; Xu, M.-H. J. Am. Chem. Soc. 2013, 135, 971. doi: 10.1021/ja3110818
-
[52]
Wang, H.; Li, Y.; Xu, M.-H. Org. Lett. 2014, 16, 3962. doi: 10.1021/ol501770q
-
[53]
Jiang, T.; Wang, Z.; Xu, M.-H. Org. Lett. 2015, 17, 528. doi: 10.1021/ol503537w
-
[54]
Zhang, X.; Xu, B.; Xu, M.-H. Org. Chem. Front. 2016, 3, 944. doi: 10.1039/C6QO00191B
-
[55]
Li, Y.; Yu, Y.-N.; Xu, M.-H. ACS Catal. 2016, 6, 661. doi: 10.1021/acscatal.5b02403
-
[56]
Zhang, Y.-F.; Chen, D.; Chen, W.-W.; Xu, M.-H. Org. Lett. 2016, 18, 2726. doi: 10.1021/acs.orglett.6b01183
-
[57]
Nishimura, T.; Noishiki, A.; Ebe, Y.; Hayashi, T. Angew. Chem., Int. Ed. 2013, 52, 1777. doi: 10.1002/anie.v52.6
-
[58]
Nishimura, T.; Ebe, Y.; Fujimoto, H.; Hayashi, T. Chem. Commun. 2013, 49, 5504. doi: 10.1039/c3cc42071j
-
[59]
Chen, Y.-J.; Chen, Y.-H.; Feng, C.-G.; Lin, G.-Q. Org. Lett. 2014, 16, 3400. doi: 10.1021/ol501464e
-
[60]
Takechi, R.; Nishimura, T. Org. Biomol. Chem. 2015, 13, 4918. doi: 10.1039/C5OB00431D
-
[61]
Yang, G.; Zhang, W. Angew. Chem., Int. Ed. 2013, 52, 7540. doi: 10.1002/anie.201302861
-
[62]
Jiang, C.; Lu, Y.; Hayashi, T. Angew. Chem., Int. Ed. 2014, 53, 9936. doi: 10.1002/anie.201406147
-
[63]
Quan, M.; Yang, G.; Xie, F.; Gridnev, I. D.; Zhang, W. Org. Chem. Front. 2015, 2, 398. doi: 10.1039/C4QO00347K
-
[64]
Álvarez-Casao, Y.; Monge, D.; Álvarez, E.; Álvarez, E.; Fernández, Lassaletta, J. M. Org. Lett. 2015, 17, 5104. doi: 10.1021/acs.orglett.5b02613
-
[65]
He, Q.; Wu, L.; Kou, X.; Butt, N.; Yang, G.; Zhang, W. Org. Lett. 2016, 18, 288. doi: 10.1021/acs.orglett.5b03458
-
[1]
-

图式2 (-)-Aurantioclavine的高效不对称合成
Scheme 2 Efficient asymmetric synthesis of (-)-aurantiocla-vine

图式6 环状N-磺酰基取代烷基酮亚胺的不对称芳基化
Scheme 6 Asymmetric arylation of cyclic N-sulfonyl aryl alkyl ketimines

图式7 简单手性膦-烯配体用于环状亚胺的不对称芳基化
Scheme 7 Simple phosphite-olefin as ligand for asymmetric arylation of cyclic ketimine

图式8 铑催化的α, β-不饱和环状酮亚胺的逐步不对称1, 4-和1, 2-加成
Scheme 8 Rh-catalyzed stepwise asymmetric 1, 4-and 1, 2-addition of α, β-unsaturated cyclic ketimines
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 54
- 文章访问数: 5578
- HTML全文浏览量: 1632
 下载:
下载:
