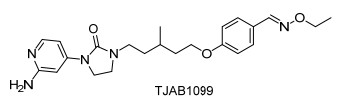
 图1
TJAB1099
Figure1.
Structure of TJAB1099
图1
TJAB1099
Figure1.
Structure of TJAB1099
引用本文:
贺万丽, 赵杨杨, 毛永红, 赵佩佩, 汪颖, 蔡岩. 肠道病毒71型抑制剂TJAB1099的实用合成方法研究[J]. 有机化学,
2017, 37(9): 2361-2368.
doi:
10.6023/cjoc201704002

Citation: He Wanli, Zhao Yangyang, Mao Yonghong, Zhao Peipei, Wang Ying, Cai Yan. Practical Synthesis of TJAB1099:An Effective Anti EV71 Inhibitor[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2361-2368. doi: 10.6023/cjoc201704002
Citation: He Wanli, Zhao Yangyang, Mao Yonghong, Zhao Peipei, Wang Ying, Cai Yan. Practical Synthesis of TJAB1099:An Effective Anti EV71 Inhibitor[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2361-2368. doi: 10.6023/cjoc201704002
肠道病毒71型抑制剂TJAB1099的实用合成方法研究
摘要:
肠道病毒71型(EV71)是目前我国手足口病的主要致病病原,EV71感染不仅造成轻症病例,还能造成重症和死亡病例,目前尚无有效治疗EV71感染的药物上市.TJAB1099是基于EV71的衣壳蛋白VP1结构设计出的有效抗病毒抑制剂分子,临床前研究证实其具有很好的成药性.主要研究了TJAB1099的有效制备方法.以2-氨基-4-溴吡啶为起始原料,经过6步化学转化,以12%的总收率得到纯度大于99%的抑制剂分子TJAB1099.合成过程中无需硅胶柱纯化操作.该路线经过多批次百克级的起始投料,收率稳定且抑制剂分子杂质含量稳定,完全可以满足其临床前实验所需的抑制剂制备量,并且也为其之后进一步的大规模生产打下基础.
English
Practical Synthesis of TJAB1099:An Effective Anti EV71 Inhibitor
Abstract:
Enterovirus 71 (EV71) is the main pathogen caused Human Hand, Foot and Mouth Disease (HFMD) in China. It not only caused mild case, but also serious case. However, no effective commercialize drugs for the treatment of HFMD were available nowadays. TJAB1099 is an effective EV71 inhibitor which was designed based on the capsid protein VP1 of EV71. The preclinical study has revealed that it owns excellent druggability. Here an practical synthesis of TJAB1099, initiated with 2-amino-4-bromide pyridine is reported. The total synthetic steps are six and its total yield is 12%. The purity of TJAB1099 is more than 99%, and silic gel chromatography is not required in the whole process. This synthetic method has been examined by hectogram level starting feeding for several times, and the total yield and the content of impurities are stable. This method could meet the need of the inhibitor amount for the preclinical study, and it could lay the foundation of further large scale synthesis.
-
Key words:
- enterovirus 71
- / inhibitor
- / synthetic method
-
手足口病(HFMD)多发于5岁以下的婴幼儿, 并且表现出广泛的临床症状, 中国一直以来都是人手足口病的高发区.引发手足口病的肠道病毒有20多种(型)[1, 2], 其中以肠道病毒71型(EV71) 及柯萨奇病毒A16 (CVA16) 最为常见, CVA16感染通常只造成轻症病例且占比例小, 而EV71感染不仅造成轻症病例, 还能造成重症和死亡病例[3, 4], 因此目前的药物研究主要是针对EV71.目前我国已有预防儿童手足口病的EV71型灭活疫苗上市[5], 然而由于病毒的高变异率, 通过接种疫苗预防EV71病毒感染仍然具有很大的挑战性.现阶段临床上针对EV71引起的手足口病感染, 一般情况下主要通过使用阿昔洛韦、利巴韦林等常用广谱抗病毒药物进行对症的支持性治疗, 而没有有效的特异性针对EV71的抗病毒药物上市[6].
作者所在的研究团队在解析了EV71的全病毒颗粒结构后[7], 根据其衣壳蛋白VP1的结构特点并借助计算机辅助药物设计, 合成筛选出了一个EV71的强效抑制剂TJAB1099(图 1)[8].其IC50值达到了25 pmol/L, 并且动物实验显示在中剂量组条件下对乳鼠的保护率接近90%, 在前期的药代动力学实验中显示出了良好的成药性, TJAB1099是目前最有可能成为特异性抗手足口病药物的候选化合物.
图1
TJAB1099
Figure1.
Structure of TJAB1099
1 结果与讨论
TJAB1099的分子结构由吡啶咪唑酮部分、作为连接的烷基链以及氧乙基肟取代的芳基三部分构成, 经过分析, 对于TJAB1099的合成主要有两种策略, 一种是以2-溴-4-氨基吡啶2为起始原料(策略a), 另一种是以2-氨基-4-溴吡啶3为起始原料(策略b), 两种策略都是经过构建吡啶咪唑酮环, 之后通过烷基链与氧乙基肟取代的芳基连接合成得到TJAB1099 (Scheme 1).
 图式 1
TJAB1099的逆合成分析
Scheme1.
Retrosynthesis of TJAB1099
图式 1
TJAB1099的逆合成分析
Scheme1.
Retrosynthesis of TJAB1099
我们前期采用策略a对TJAB1099进行合成[9], 以2-溴-4-氨基吡啶(2)为起始原料, 首先与2-氯乙基异氰酸酯进行无溶剂反应, 之后原位关环得到吡啶咪唑烷酮中间体4, 以该中间体为起始原料, 我们先后分别采用了两种方法进行合成, 第一种方法是中间体4与1, 5-二溴-3-甲基戊烷反应得到中间体5, 接着与对羟基苯甲醛反应得到中间体6, 随后在醋酸钯的催化下与氨水进行封管反应得到中间体7, 最后与羟胺缩合得到TJAB1099 (Scheme 2, path a), 该路线由于在吡啶环上的溴转化氨基这一步反应的收率过低, 通过大量的条件优化, 产率未能得到明显改善, 最终未被采纳.第二种方法是先将中间体4上的溴原子转化为叠氮基团, 再与1, 5-二溴-3-甲基戊烷反应得到中间体9, 之后将叠氮基团还原为氨基得到中间体10, 最后与中间体11反应得到TJAB1099 (Scheme 2, path b), 该方法的总收率为10%, 经过多次投料收率稳定, 其制备量满足了我们前期进行药效学方面实验对化合物的需求, 但是该方法仍然有着固有缺点, 第一是反应用到的2-氯乙基异氰酸酯毒性较大, 大规模制备时容易对环境和操作人员造成危害, 第二是反应中用到了大量易爆的叠氮化钠, 限制了之后的放大生产, 第三还原叠氮基团时会产生大量的锡盐废渣, 不易后处理且很容易造成最终产品的重金属超标, 因此, 随着临床前实验对化合物的需求量增加, 以及考虑到会对之后药物申报方面带来不利的影响, 最终放弃了这个合成方法.
 图式 2
前期采取的合成策略
Scheme2.
Previous synthetic strategy
图式 2
前期采取的合成策略
Scheme2.
Previous synthetic strategy
为了克服以上路线的缺点, 我们设计开发了以2-氨基-4-溴吡啶(3)为起始原料(Scheme 1, 策略b), 利用钯催化偶联的方法来合成TJAB1099的方法.其合成始于用对甲氧基苄基(PMB)对2氨基-4-溴吡啶(3)的氨基进行保护得到中间体化合物12[10], 随后与单乙酰基咪唑酮13进行钯催化的偶联反应, 得到吡啶咪唑烷酮中间体14, 然后在碱性条件下脱去乙酰基保护得到中间体化合物15, 之后在碱性条件下与1, 5-二溴-3-甲基-戊烷反应得到中间体化合物16, 随后脱去PMB保护基得到中间体化合物10, 最后在碱性条件下与化合物11反应得到TJAB1099 (Scheme 3), 该路线避免了之前所采用制备方法存在的问题, 满足了我们在进行临床前实验对TJAB1099的化合物需求量, 也利于之后的放大生产.
 图式 3
TJAB1099的实用合成方法
Scheme3.
Pactical synthesis of TJAB1099
图式 3
TJAB1099的实用合成方法
Scheme3.
Pactical synthesis of TJAB1099
1.1 氨基保护基的选择以及偶联反应条件的优化
该路线的关键步骤在于通过偶联反应形成C-N键来构建吡啶咪唑烷酮14这个关键中间体, 我们在优化偶联反应条件的同时, 也考察了氨基保护基的影响.我们首先选择甲酸酯作为氨基保护基, 铜盐作为催化剂进行反应, 但是未得到预期产物, 原料全部分解(表 1, Entries 1, 2).之后我们选择Pd2dba3作为催化剂, BINAP作为配体进行反应, 发现反应可以进行, 但是产率极低(表 1, Entries 3~6), 当将配体换为Xantphos, Cs2CO3作为碱时, 反应可以23%的分离收率得到预期的产物14(表 1, Entry 7), 随后我们通过更换不同的碱、催化剂以及溶剂以期能够进一步提高收率, 但效果不理想(表 1, Entries 8~11).我们考虑可能2-氨基-4-溴吡啶(3)上的氨基保护基会对偶联反应造成影响, 所以将甲酸酯保护基换为了叔丁氧羰基进行考察, 但是反应却未得到预期的产物(表 1, Entries 12~14).令我们惊喜的是, 当将氨基保护基换为对甲氧基苄基时, 以Pd2dba3作为催化剂, Xantphos为配体, Cs2CO3作为碱时, 反应可以60%的分离收率得到预期产物(表 1, Entry 15), 随后的条件优化却未能进一步提高收率(表 1, Entries 16~19), 当将反应温度或催化剂/配体的量降低时, 反应收率也会降低(表 1, Entries 20, 21).因此, 反应优化后的条件为:以对甲氧基苄基作为2-氨基-4-溴吡啶(3)上的氨基保护基, 以10 mol%的Pd2dba3作为催化剂, 11 mol%的Xantphos为配体, 1.5 equiv.的Cs2CO3作为碱, 甲苯作为溶剂, 反应温度为90 ℃.在后续的放大实验中, 我们发现产物可以通过乙酸乙酯/石油醚进行重结晶而得到纯化, 无需再用硅胶柱纯化.
 表 1
氨基保护基的选择以及偶联反应条件的优化a
Table 1.
Selection of protect group of amino and the condition optimization of coupling reaction
表 1
氨基保护基的选择以及偶联反应条件的优化a
Table 1.
Selection of protect group of amino and the condition optimization of coupling reaction

Entry R Cat. Ligand Base Solvent Yieldb/% 1 
CuI N, N'-Dimethylethylenediamine K2CO3 n-BuOH ND 2 CuI N, N'-Dimethylethylenediamine Cs2CO3 Dioxnae ND 3 Pd2dba3 BINAP K3PO4 Toluene Trace 4 Pd2dba3 BINAP Cs2CO3 Toluene Trace 5 Pd2dba3 BINAP t-BuONa Toluene ND 6 Pd2dba3 Johnphos Cs2CO3 Toluene Trace 7 Pd2dba3 Xantphos Cs2CO3 Toluene 23 8 Pd2dba3 Xantphos K3PO4 Toluene Trace 9 Pd2dba3 Xantphos t-BuONa Toluene ND 10 Pd(OAc)2 Xantphos Cs2CO3 Toluene 12 11 Pd(OAc)2 Xantphos Cs2CO3 Dioxane 4 12 Pd2dba3 Xantphos Cs2CO3 Toluene ND 13 Pd2dba3 Xantphos K3PO4 Toluene ND 14 Pd2dba3 Xantphos t-BuONa Toluene ND 15 Pd2dba3 Xantphos Cs2CO3 Toluene 60 16 Pd2dba3 Xantphos K3PO4 Toluene 26 17 Pd2dba3 Xantphos t-BuONa Toluene 8 18 Pd2dba3 Johnphos Cs2CO3 Toluene 47 19 Pd2dba3 Xantphos Cs2CO3 Dioxane 31 20 Pd2dba3 Xantphos Cs2CO3 Toluene 53c 21 Pd2dba3 Xantphos Cs2CO3 Toluene 37d a一般反应条件:将200 mg化合物12以及1.2 equiv.的化合物13置于5 mL甲苯中, 加入10 mol%的催化剂、11 mol%的配体以及1.5 equiv.的碱, 氮气保护, 90℃反应15 h. b硅胶柱分离收率. c反应温度为60 ℃. d催化剂的加入量为5 mol%, 配体的加入量为6 mol%. ND, no desired. 1.2 吡啶咪唑烷酮与1, 5-二溴-3-甲基-戊烷反应条件的优化
接下来我们筛选优化了吡啶咪唑烷酮(中间体15)与1, 5-二溴-3-甲基-戊烷[11]的反应条件.我们首先筛选了不同的碱, 发现叔丁醇钾、氢氧化钾以及碳酸钾均无法使原料发生转化(表 2, Entries 1~3), 即便在加入相转移催化剂的情况下, 反应也无法发生(表 2, Entry 4).当将碱换为脱氢能力更强的氢化钠时, 反应顺利进行, 并以50%的收率得到目标产物(表 2, Entry 5).将氢化钠的用量增加, 发现会对反应不利, 收率会降至41%(表 2, Entry 6), 提升1, 5-二溴-3-甲基-戊烷的量会使反应的收率增加到67%(表 2, Entry 7), 但继续增加1, 5-二溴-3-甲基-戊烷的量, 却无法进一步提升收率, 收率反而会有所下降(表 2, Entry 8), 降低氢化钠的用量也会使得反应收率下降(表 2, Entry 9).因此, 该步反应的最优条件确定为: 1, 5-二溴-3-甲基-戊烷的投料量为2 equiv., 1.5 equiv.氢化钠作为碱, N, N-二甲基甲酰胺(DMF)作为溶剂, 室温反应.此外, 我们在条件优化的过程中, 发现总是有1, 5-二溴-3-甲基-戊烷的两侧都连接有吡啶咪唑烷酮的副产物16', 无论条件如何优化, 都难以避免, 但是该副产物可以通过乙酸乙酯萃取的方法有效除去, 在之后的放大过程中, 该步反应的产物粗品可以不经纯化直接用于后续反应投料.
表 2
吡啶咪唑烷酮与1, 5-二溴-3-甲基-戊烷反应条件的优化a
Table 2.
Condition optimization of the reaction of pyridine imidazolidinone and 1, 5-dibromide-3-methyl-pentane

Entry x/equiv. Base y/equiv. Solvent Temp./℃ Yieldb/% 1 1.5 t-BuOK 1.5 THF 60 NR 2 1.5 KOH 1.5 Toluene 90 NR 3 1.5 K2CO3 1.5 Acetone 60 NR 4c 1.5 K2CO3 1.5 Acetone 60 NR 5 1.5 NaH 1.5 DMF 25 50 6 1.5 NaH 2 DMF 25 41 7 2 NaH 1.5 DMF 25 67 8 2.5 NaH 1.5 DMF 25 63 9 2 NaH 1.1 DMF 25 52 a一般反应条件:将500 mg化合物15以及x equiv.的1, 5-二溴-3-甲基-戊烷加入到10 mL DMF中, 体系于冰水浴条件下冷却, 之后逐渐加入y equiv.的碱, 加入完毕后搅拌10 min, 之后升至相应的温度反应3 h. b硅胶柱分离收率. c反应过程中加入了1 equiv.的四丁基硫酸氢铵(TBAB). NR, no reaction. 1.3 TJAB1099的合成
中间体16在三氟乙酸存在的条件下脱去氨基上的PMB保护基得到中间体10, 它与1.6 equiv.中间体11, 在5 equiv.碳酸钾作用下, 65 ℃反应8 h, 得到TJAB1099的粗品, 经过乙醇洗涤, 乙酸乙酯重结晶, 可以得到纯度大于99%的产品, 该步反应产物结晶后的收率为56% (Scheme 4).其中中间体11可以通过对羟基苯甲醛与氧乙基羟肟盐酸盐在乙酸钠的作用下, 室温反应得到[12].
 图式 4
TJAB1099的合成
Scheme4.
Synthesis of TJAB1099
图式 4
TJAB1099的合成
Scheme4.
Synthesis of TJAB1099
1.4 TJAB1099的合成成本的初步核算对比
工艺路线合成设计中的成本是一个比较重要的考量要素, 我们将之前采用的合成策略a与目前采用的合成策略b中使用的主要原料成本做了初步的罗列对比, 以制备10 g纯度大于99%的TJAB1099为例进行核算, 所需主要试剂的用量以及价格如表 3和表 4所示, 可以看出采用策略b进行制备所需主要试剂的成本要略低于之前的合成策略.
表 3
采用策略a制备10 g纯度大于99%的TJAB1099所需主要试剂的成本
Table 3.
Cost of main reagents required in the preparation of 10 g of TJAB1099 ( > 99% purity) with the use of strategy
原料名称 采购单价/元 采购包装规格/g 实际用量/g 总价/元 供应商 2-溴-4-氨基吡啶 576.00 25 40.4 930.81 北京伊诺凯科技有限公司 2-氯乙基异氰酸酯 1742.00 100 100 1742.00 百灵威科技有限公司 叠氮化钠 160.00 50 33.4 106.88 西亚试剂 氯化亚锡二水合物 59.00 100 35.3 20.83 百灵威科技有限公司 价格总计/元 2800.52
表 4
采用策略b制备10 g纯度大于99%的TJAB1099所需主要试剂的成本
Table 4.
Cost of main reagents required in the preparation of 10 g TJAB1099 ( > 99% purity) with the use of strategy
原料名称 采购单价/元 采购包装规格 实际用量/g 总价/元 供应商 2-氨基-4-溴吡啶 540.00 25 g 31.8 686.88 北京伊诺凯科技有限公司 4-甲氧基苄氯 499.00 100 mL 91.8 398.33 天津希恩思奥普德科技有限公司 Pd2(dba)3 756.00 10 g 14.4 1088.64 北京伊诺凯科技有限公司 Xantphos 432.00 25 g 10.0 172.80 北京伊诺凯科技有限公司 N-乙酰基-2-咪唑烷酮 283.10 25 g 24.2 274.00 安耐吉化学 价格总计/元 2620.65 2 结论
我们成功设计开发了以2-氨基-4-溴吡啶为起始原料, 利用钯催化偶联的方法来合成TJAB1099的方法, 该方法避免了之前的制备方法存在的问题, 该合成路线均不需要硅胶柱层析, 所有的中间体经过适当洗涤或结晶纯化后直接进行下一步投料, 且终产物TJAB1099通过重结晶可以达到99%以上纯度, 该路线已进行了多次百克级的起始放大投料, 产率较为稳定, 同时满足了我们在进行临床前实验对TJAB1099的化合物需求量, 也利于之后的放大生产.
3 实验部分
3.1 仪器与试剂
仪器:核磁共振仪Bruker Plus 400M型, 质谱仪Waters 3100 Mass Detctor, 分析型液相色谱仪Hitachi L-2000型, 旋转蒸发仪YRE-2000A型, UAT-02暗箱式紫外分析仪, WRX-4显微熔点仪.
试剂: 1, 5-二溴-3-甲基-戊烷与化合物11为自制; 其余所用试剂均为分析纯, 氮气为高纯氮, 柱层析所用硅胶(200~300目)和GF254薄层硅胶板均为青岛海洋化工厂生产.
3.2 实验方法
3.2.1 4-溴-N, N-双(4-甲氧基苄基)吡啶-2-氨(12)的制备
将100 g的2-氨基-4-溴吡啶溶于2 L的DMF中, 将体系置于低温反应釜中冷却, 使温度降至-5 ℃, 之后将89.2 g质量分数为60%的NaH分批加入到上述反应体系中, 加料完毕后在氮气保护下保温搅拌0.5 h, 之后向反应体系缓慢滴加4-甲氧基苄氯(PMBCl) 288.6 g, 滴加完毕后自然恢复至室温, 反应7 h后, 薄层色谱(TLC)监测原料基本反应完全, 将反应体系控制在30 ℃以下, 加入2.5 L H2O将反应淬灭, 有大量固体析出, 用真空隔膜泵进行抽滤, 收集析出的固体, 滤饼用1.5 L H2O进行洗涤, 之后干燥得到204.5 g黄色固体, 即中间体12, 产率85.6%. m.p. 126.2~126.9 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 8.03 (d, J=5.3 Hz, 1H), 7.15 (d, J=8.6 Hz, 4H), 6.87 (d, J=8.7 Hz, 4H), 6.75 (dd, J=5.4, 1.5 Hz, 1H), 6.67 (d, J=1.5 Hz, 1H), 4.69 (s, 4H), 3.82 (s, 6H); HR-ESI-MS calcd for C21H22BrN2O2 [M+H]+ 414.3150, found 414.3102.
3.2.2 1-乙酰基-3-(2-(双(4-甲氧基苄基)氨基)吡啶-4)-咪唑烷-2-酮(14)的制备
将170.7 g的化合物12加入到3 L的甲苯中, 之后依次向上述体系中加入55.2 g的N-乙酰基咪唑烷酮、175.4 g Cs2CO3、20.6 g Pd2(dba)3和22.8 g Xantphos, 氮气保护, 加热体系使内温升至90 ℃, 加热回流反应15 h, TLC监测原料反应完全, 停止反应, 待体系降至室温, 将体系过滤, 收集滤液并旋去溶剂, 剩余物溶于乙酸乙酯中, 滤饼用1 L乙酸乙酯淋洗, 之后合并所有的乙酸乙酯相再用1.5 L饱和NaCl溶液洗涤一次, 收集有机相并用无水硫酸钠干燥, 之后将有机相减压浓缩除去溶剂, 得259 g棕色油状物, 对产物粗品进行重结晶:将259 g棕色油状物用250 mL的乙酸乙酯加热至75 ℃, 向体系中缓慢加入250 mL的石油醚, 体系析出大量白色固体, 自然降温至25 ℃, 抽滤, 收集固体, 并将所得固体进行真空干燥得104.8 g白色固体, 即为中间体14, 产率为53.7%. m.p. 132.5~133.1 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 8.17 (d, J=5.8 Hz, 1H), 7.18 (d, J=8.6 Hz, 4H), 6.88~6.90 (m, 1H), 6.83~6.88 (m, 4H), 6.68~6.71 (m, 1H), 4.73 (s, 4H), 3.87~3.95 (m, 2H), 3.81 (s, 6H), 3.69~3.77 (m, 2H), 2.56 (s, 3H); IR (KBr) ν: 3259.34, 3167.51, 1738.1, 1654.23, 1609.55, 1598.37, 1511.48, 1500.06, 1477.01, 1406.9, 1382.01, 1365.33, 1349.41, 1307.6, 1303.5, 1290.6, 1264.8, 1247.29, 1219.37, 1176.11, 1170.13, 606.66 cm-1; HR-ESI-MS calcd for C26H29N4O4 [M+H]+ 461.5340, found 461.5304.
3.2.3 1-(2-(双(4-甲氧基苄基)氨基)吡啶-4) 咪唑烷-2-酮的制备(15)的制备
将104.8 g化合物14分散到900 mL的MeOH中, 再将80 g的K2CO3加入到体系中, 室温反应3 h, TLC监测至原料反应完全, 向体系中加入450 mL的水, 搅拌30 min后, 将体系直接过滤, 滤饼用450 mL的水洗涤, 后将所得固体真空干燥, 得90.4 g类白色固体, 即为中间体15, 产率为92%. m.p. 155.2~155.6 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 8.11 (d, J=5.8 Hz, 1H), 7.17~7.21 (m, 4H), 6.87~6.89 (m, 1H), 6.83~6.87 (m, 4H), 6.71~6.74 (m, 1H), 4.72 (s, 4H), 4.68 (s, 1H), 3.81 (s, 6H), 3.74~3.80 (m, 2H), 3.49~3.58 (m, 2H); IR (KBr) ν: 3238.77, 3166.25, 2904.66, 1721.67, 1597.58, 1548.44, 1511.2, 1494.74, 1478.78, 1030.82, 811.3 cm-1. HR-ESI-MS calcd for C24H27N4O3 [M+H]+ 419.4970, found 419.4906.
3.2.4 1-(2-(双(4-甲氧基苄基)氨基)吡啶-4)-3-(5-溴-3-甲基戊基)咪唑烷-2-酮(16)的制备
将44.4 g化合物15溶于740 mL的DMF中, 将21.2 g 3-甲基-1, 5-二溴戊烷加入上述体系, 氮气保护下将体系置于低温反应釜中, 并降温至0 ℃, 之后向体系中分批加入5.5 g质量分数为60%的NaH, 加料完毕后保温反应30 min, 之后自然升温至25 ℃反应, 反应3 h后, TLC监测原料反应完全, 将体系冷却至0 ℃, 之后向反应体系中加入500 mL的水, 体系用乙酸乙酯萃取(300 mL×3), 合并有机相, 有机相依次用300 mL H2O和300 mL饱和NaCl溶液洗涤, 收集有机相, 之后用无水Na2SO4干燥, 减压浓缩除去溶剂, 得82 g棕黄色油状物, 即为化合物16粗品, 不经纯化可直接用于下一步反应投料. 1H NMR (400 MHz, Chloroform-d) δ: 8.09 (d, J=5.9 Hz, 1H), 7.15~7.21 (m, 4H), 6.89 (dd, J=5.9, 1.9 Hz, 1H), 6.81~6.88 (m, 4H), 6.73 (d, J=1.9 Hz, 1H), 4.71 (s, 4H), 3.81 (s, 6H), 3.63~3.70 (m, 2H), 3.24~3.52 (m, 6H), 1.87~1.96 (m, 1H), 1.69~1.81 (m, 2H), 1.64~1.69 (m, 1H), 1.35~1.42 (m, 1H), 0.98 (d, J=6.1 Hz, 3H); IR (KBr) ν: 3435.4, 2927.9, 1708.53, 1608.76, 1509.88, 1477.05, 1422.99, 1384.43, 816.48 cm-1; HR-ESI-MS calcd for C30H38BrN4O3 [M+H]+ 582.5550, found 582.5508.
3.3.5 1-(2-氨基吡啶-4-基)-3-(5-溴-3-甲基戊基)咪唑烷-2-酮(10)的制备
将82 g化合物16的粗品溶于660 mL的二氯甲烷(DCM)中, 后将328 mL三氟乙酸加入到体系中, 室温反应3 h, TLC监测原料反应完全.直接将反应体系减压浓缩除去溶剂, 后向体系中加入饱和NaHCO3溶液, 将pH调节为7, 之后向体系中加入400 mL的乙酸乙酯进行萃取、分液, 收集乙酸乙酯相, 水相再次用乙酸乙酯萃取(200 mL×2), 合并有机相, 并用300 mL的饱和NaCl溶液洗涤, 之后用无水Na2SO4干燥, 减压浓缩除去溶剂, 得72 g黄色粘稠物, 对产物粗品进行重结晶:向体系中加入50 mL的乙酸乙酯加热至75 ℃, 向其中缓慢加入75 mL甲基叔丁基醚, 有白色固体析出, 后自然恢复至25℃, 过滤、收集固体, 真空干燥得19.5 g白色固体, 即中间体10, m.p. 116.5~117.2 ℃, 与上步反应收率合计为54%. 1H NMR (400 MHz, Chloroform-d) δ: 7.96 (d, J=5.9 Hz, 1H), 6.93~6.98 (m, 1H), 6.75 (dd, J=6.1, 1.9 Hz, 1H), 4.37~4.49 (m, 2H), 3.80 (t, J=8.1 Hz, 2H), 3.51~3.59 (m, 2H), 3.43~3.50 (m, 2H), 3.27~3.42 (m, 2H), 1.89~1.99 (m, 1H), 1.72~1.82 (m, 2H), 1.59~1.69 (m, 1H), 1.37~1.47 (m, 1H), 1.01 (d, J=6.0 Hz, 3H); IR (KBr) ν: 3419.94, 3295.1, 3158.42, 2964.13, 2926.95, 2371.29, 1700.87, 1691.3, 1639.83, 1605.56, 1547.82, 1483.84, 1433.89, 1297.29, 1277.34, 824.2 cm-1; HR-ESI-MS calcd for C14H22BrN4O [M+H]+ 342.2530, found 342.2510.
3.2.6 (E)-4-((5-(3-(2-氨基吡啶-4)-2-氧代咪唑烷-1)-3-甲基戊基)氧基)苯甲醛-O-乙基肟(TJAB1099) 的制备
将12.4 g化合物10和6.9 g的化合物11、17.4 g的K2CO3加入到86 mL CH3CN中, 65 ℃加热回流, 反应6 h后, TLC监测原料反应完毕, 停止反应, 待体系温度降至室温, 过滤除去体系中的固体, 收集滤液, 滤饼用DCM洗涤, 合并有机相并减压浓缩除去溶剂, 得18.7 g黄色固体.对所得产物粗品用乙醇进行洗涤后进行重结晶:向体系中加入10 mL乙酸乙酯稀释, 75 ℃加热回流, 后缓慢加入乙酸乙酯直至体系刚刚澄清透明, 自然降至室温, 有白色固体析出, 减压抽滤, 收集滤饼, 并将所得固体真空干燥, 得白色固体粉末8.6 g, 收率为56%.即TJAB1099. m.p. 135.4~136.9 ℃; 1H NMR (400 MHz, Chloroform-d) δ: 8.04 (s, 1H), 7.96 (d, J=5.9 Hz, 1H), 7.47~7.55 (m, 2H), 6.9~6.98 (m, 1H), 6.84~6.91 (m, 2H), 6.74 (dd, J=6.0, 2.0 Hz, 1H), 4.38 (s, 2H), 4.18~4.26 (m, 2H), 4.00~4.10 (m, 2H), 3.69~3.81 (m, 2H), 3.45~3.54 (m, 2H), 3.29~3.43 (m, 2H), 1.74~1.93 (m, 2H), 1.63~1.74 (m, 2H), 1.41~1.51 (m, 1H), 1.34 (t, J=7.1 Hz, 3H), 1.05 (d, J=6.4 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ: 160.22, 159.36, 156.93, 148.65, 148.47, 147.95, 128.39, 124.98, 114.61, 102.79, 94.74, 69.54, 65.89, 41.77, 41.54, 41.34, 35.92, 34.16, 27.50, 19.46, 14.63; IR (KBr) ν: 3418.06, 3294.25, 3157.71, 2931.26, 2879.04, 1699.9, 1689.97, 1641.06, 1606.71, 1547.88, 1506.67, 1482.88, 1376.71, 1250.65, 1055.84, 961.97, 825.75 cm-1; HR-MS calcd for C23H32N5O3 [M+H]+ 426.5330, found 426.5303.
-
-
[1]
(a) McMinn, P. C. FEMS Microbio. Rev. 2002, 26, 91.
(b) Shih, S. R.; Chen, S. J.; Hakimelahi, G. H.; Liu, H. J.; Tseng, C. T.; Shia, K. S. Med. Res. Rev. 2004, 24, 449. -
[2]
桂娟娟, 刘志芳, 华启航, 董长征, 遗传, 2015, 37, 426. http://www.ebiotrade.com/emagazine/content/1/2015_5_37_5/21CE8BB3-02C2-45F5-AC59-AE6EBD1671D2/pdf/426.pdf
-
[3]
(a) Chang, L.Y.; Lin, T. Y.; Hsu, K. H.; Huang, Y. C.; Lin, K. L.; Hsueh, C.; Shih, S. R.; Ning, H. C.; Hwang, M. S.; Wang, H. S.; Lee, C. Y. Lancet 1999, 354, 1682.
(b) Chan, K. P.; Goh, H. T.; Chong, C. Y.; Teo, E. S.; Lau, G.; Ling, A. E. Emerging Infect. Dis. 2003, 9, 78.
(c) Huang, K. Y.; Zhang, X.; Chung, P. H.; Tsao, K. C.; Lin, T. S.; Su, L. H., Chiu, C. H. Scand. J. Infect. Dis. 2008, 40, 571. -
[4]
(a) Tan, X.; Huang, X.; Zhu, S.; Chen, H.; Yu, Q.; Wang, H.; Huo, X.; Zhou, J.; Wu, Y.; Yan, D. PLos One 2011, 6, e25662.
(b) Zeng, D. B.; Ma, Y. Y.; Zhang, R.; Nie, Q. D.; Cui, Z. J.; Wang, Y. X.; Shang, L. Q.; Yin, Z. Bioorg. Med. Chem. 2016, 26, 1762. -
[5]
Li, R. C.; Liu, L. D.; Mo, Z. J.; Wang, X. Y.; Xia, J. L.; Liang, Z. L.; Zhang, Y.; Li, Y. P.; Mao, Q. Y.; Wang, J. J.; Jiang, L.; Dong, C. H.; Che, Y. C.; Huang, T.; Jiang, Z. W.; Xie, Z. P.; Wang, L. C.; Liao, Y.; Liang, Y.; Nong, Y.; Liu, J. S.; Zhao, H. L.; Na, R. X.; Guo, L.; Pu, J.; Yang, E.; Sun, L.; Cui, P. F.; Shi, H. J.; Wang, J. Z.; Li, Q. H. New Engl. J. Med. 2014, 370, 829. doi: 10.1056/NEJMoa1303224
-
[6]
郭辉, 贾贻红, 中国医药指南, 2009, 7, 66. doi: 10.3969/j.issn.1671-8194.2009.01.048Guo, H.; Jia, Y. H. Guid. Chin. Med. 2009, 7, 66(in Chinese). doi: 10.3969/j.issn.1671-8194.2009.01.048
-
[7]
Wang, X. X.; Peng, W.; Ren, J. S.; Hu, Z. Y.; Xu, J. W.; Lou, Z. Y.; Li, X. M.; Yin, W. D.; Shen, X. L.; Porta, C.; Walter, T. S.; Evans, G.; Axford, D.; Owen, R.; Rowlands, D.; Wang, J. Z.; Stuart, D. I.; Fry, E. E.; Rao, Z. H. J. Nat. Struct. Mol. Biol. 2012, 19, 424. doi: 10.1038/nsmb.2255
-
[8]
De Colibus, L.; Wang, X. X.; Spyrou, J. A. B.; Kelly, J.; Ren, J. S.; Grimes, J.; Puerstinger, G.; Stonehouse, N.; Walter, T. S.; Hu, Z. Y.; Wang, J. Z.; Li, X. M.; Peng, W.; Rowlands, D. J. Fry, E. E.; Rao, Z. H.; Stuart, D. I. J. Nat. Struct. Mol. Biol. 2014, 21, 282. doi: 10.1038/nsmb.2769
-
[9]
Rao, Z. H.; Yang, C.; Cai, Y.; Guo, Y.; Li, S.; Wang, Y.; Mao, Y. H.; Zhao, P. P.; He, W. L.; Zhao, Y. Y.; Liu, Y. H.; Meng, F. F. CN 201610315888.7, 2016. http://www.sibcb.ac.cn/ePI.asp?id=137
-
[10]
Lin, L. S.; Cui, M. X.; Hu, B.; Hao, J. L.; Chen, Z. X. WO 2014022528[Chem. Abstr. 2014, 160, 265016].
-
[11]
Avis, T. J.; Boulanger, R. R.; Belanger, R. R. J. Chem. Ecol. 2000, 26, 987. doi: 10.1023/A:1005464326573
-
[12]
Chern, J. H.; Lee, C. C.; Chang, C. S.; Lee, Y. C.; Tai, C. L.; Lin, Y. T.; Shia, K. S.; Lee, C. Y.; Shih, S. R. Bioorg. Med. Chem. Lett. 2004, 14, 5051. doi: 10.1016/j.bmcl.2004.07.084
-
[1]
-
表 1 氨基保护基的选择以及偶联反应条件的优化a
Table 1. Selection of protect group of amino and the condition optimization of coupling reaction
Entry R Cat. Ligand Base Solvent Yieldb/% 1 CuI N, N'-Dimethylethylenediamine K2CO3 n-BuOH ND 2 CuI N, N'-Dimethylethylenediamine Cs2CO3 Dioxnae ND 3 Pd2dba3 BINAP K3PO4 Toluene Trace 4 Pd2dba3 BINAP Cs2CO3 Toluene Trace 5 Pd2dba3 BINAP t-BuONa Toluene ND 6 Pd2dba3 Johnphos Cs2CO3 Toluene Trace 7 Pd2dba3 Xantphos Cs2CO3 Toluene 23 8 Pd2dba3 Xantphos K3PO4 Toluene Trace 9 Pd2dba3 Xantphos t-BuONa Toluene ND 10 Pd(OAc)2 Xantphos Cs2CO3 Toluene 12 11 Pd(OAc)2 Xantphos Cs2CO3 Dioxane 4 12 Pd2dba3 Xantphos Cs2CO3 Toluene ND 13 Pd2dba3 Xantphos K3PO4 Toluene ND 14 Pd2dba3 Xantphos t-BuONa Toluene ND 15 Pd2dba3 Xantphos Cs2CO3 Toluene 60 16 Pd2dba3 Xantphos K3PO4 Toluene 26 17 Pd2dba3 Xantphos t-BuONa Toluene 8 18 Pd2dba3 Johnphos Cs2CO3 Toluene 47 19 Pd2dba3 Xantphos Cs2CO3 Dioxane 31 20 Pd2dba3 Xantphos Cs2CO3 Toluene 53c 21 Pd2dba3 Xantphos Cs2CO3 Toluene 37d a一般反应条件:将200 mg化合物12以及1.2 equiv.的化合物13置于5 mL甲苯中, 加入10 mol%的催化剂、11 mol%的配体以及1.5 equiv.的碱, 氮气保护, 90℃反应15 h. b硅胶柱分离收率. c反应温度为60 ℃. d催化剂的加入量为5 mol%, 配体的加入量为6 mol%. ND, no desired.  下载: 导出CSV
下载: 导出CSV
表 2 吡啶咪唑烷酮与1, 5-二溴-3-甲基-戊烷反应条件的优化a
Table 2. Condition optimization of the reaction of pyridine imidazolidinone and 1, 5-dibromide-3-methyl-pentane
Entry x/equiv. Base y/equiv. Solvent Temp./℃ Yieldb/% 1 1.5 t-BuOK 1.5 THF 60 NR 2 1.5 KOH 1.5 Toluene 90 NR 3 1.5 K2CO3 1.5 Acetone 60 NR 4c 1.5 K2CO3 1.5 Acetone 60 NR 5 1.5 NaH 1.5 DMF 25 50 6 1.5 NaH 2 DMF 25 41 7 2 NaH 1.5 DMF 25 67 8 2.5 NaH 1.5 DMF 25 63 9 2 NaH 1.1 DMF 25 52 a一般反应条件:将500 mg化合物15以及x equiv.的1, 5-二溴-3-甲基-戊烷加入到10 mL DMF中, 体系于冰水浴条件下冷却, 之后逐渐加入y equiv.的碱, 加入完毕后搅拌10 min, 之后升至相应的温度反应3 h. b硅胶柱分离收率. c反应过程中加入了1 equiv.的四丁基硫酸氢铵(TBAB). NR, no reaction.
下载: 导出CSV
表 3 采用策略a制备10 g纯度大于99%的TJAB1099所需主要试剂的成本
Table 3. Cost of main reagents required in the preparation of 10 g of TJAB1099 ( > 99% purity) with the use of strategy
原料名称 采购单价/元 采购包装规格/g 实际用量/g 总价/元 供应商 2-溴-4-氨基吡啶 576.00 25 40.4 930.81 北京伊诺凯科技有限公司 2-氯乙基异氰酸酯 1742.00 100 100 1742.00 百灵威科技有限公司 叠氮化钠 160.00 50 33.4 106.88 西亚试剂 氯化亚锡二水合物 59.00 100 35.3 20.83 百灵威科技有限公司 价格总计/元 2800.52
下载: 导出CSV
表 4 采用策略b制备10 g纯度大于99%的TJAB1099所需主要试剂的成本
Table 4. Cost of main reagents required in the preparation of 10 g TJAB1099 ( > 99% purity) with the use of strategy
原料名称 采购单价/元 采购包装规格 实际用量/g 总价/元 供应商 2-氨基-4-溴吡啶 540.00 25 g 31.8 686.88 北京伊诺凯科技有限公司 4-甲氧基苄氯 499.00 100 mL 91.8 398.33 天津希恩思奥普德科技有限公司 Pd2(dba)3 756.00 10 g 14.4 1088.64 北京伊诺凯科技有限公司 Xantphos 432.00 25 g 10.0 172.80 北京伊诺凯科技有限公司 N-乙酰基-2-咪唑烷酮 283.10 25 g 24.2 274.00 安耐吉化学 价格总计/元 2620.65
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 1917
- HTML全文浏览量: 189
 下载:
下载:
