 图式1
冬凌草甲素C(1) 和C(14) 衍生物合成路线
图式1.
Synthetic route of C(1) and C(14) derivatives of oridonin
图式1
冬凌草甲素C(1) 和C(14) 衍生物合成路线
图式1.
Synthetic route of C(1) and C(14) derivatives of oridonin
引用本文:
戴一, 仲飞. 冬凌草甲素的结构修饰与生物活性研究进展[J]. 有机化学,
2017, 37(7): 1701-1713.
doi:
10.6023/cjoc201702011

Citation: Dai Yi, Zhong Fei. Advances in the Study of Structural Modification and Biological Activities of Oridonin[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1701-1713. doi: 10.6023/cjoc201702011
Citation: Dai Yi, Zhong Fei. Advances in the Study of Structural Modification and Biological Activities of Oridonin[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1701-1713. doi: 10.6023/cjoc201702011
冬凌草甲素的结构修饰与生物活性研究进展
English
Advances in the Study of Structural Modification and Biological Activities of Oridonin
Abstract:
Oridonin, an ent-kaurane diterpenoid, is found in the Chinese herb Rabdosia rubescens and some related species, and has various biological activities such as anti-tumor, anti-microbial, anti-inflammatory, and so on.This review provides an overview of the multifunctional effects of the structural modification of oridonin since 2000, suggesting that it may be effective choice for improving pharmacological activities.
-
Key words:
- oridonin
- / structural modification
- / anti-cancer activity
- / anti-mycobacterial activity
-
冬凌草甲素(1)为对映贝壳杉烷类二萜, 来源于中草药冬凌草(Rabdosia rubescens)及相关植物.冬凌草甲素具有多种药理活性如抗炎、抗微生物和抗肿瘤等作用, 尤其是抗肿瘤作用受到极大的关注[1], 如由冬凌草等8种中草药组成的PC-SPES复方临床有效治疗前列腺癌, 其中冬凌草甲素起着非常关键的作用[2].冬凌草甲素的抗肿瘤机制复杂, 涉及细胞周期阻滞、凋亡、自噬、癌蛋白调节、ROS聚集、MAPKs和PI3K/Akt信号通路调节及MicroRNAs正常表达改变等[3].另外, 冬凌草甲素在非癌症疾病治疗方面也具有一定的潜力[4].鉴于冬凌草甲素多样的药理作用, 其应用性开发值得深入研究, 但冬凌草甲素也存在不足, 如水溶性差、药理作用温和及没有靶向性等, 针对上述不足, 对冬凌草甲素进行结构修饰是有效的改善方法.结构修饰对于优化药物结构、改善药物体内过程及提高活性等也起到积极作用[5], 多年来冬凌草甲素结构修饰国内外研究活跃, 取得了一定成果, 本文通过检索web of science和SciFinder对冬凌草甲素结构修饰进行综述, 以期为冬凌草甲素的进一步开发提供参考.
1 冬凌草甲素结构修饰与抗肿瘤活性
1.1 C(1) 位或(和)C(14) 位修饰
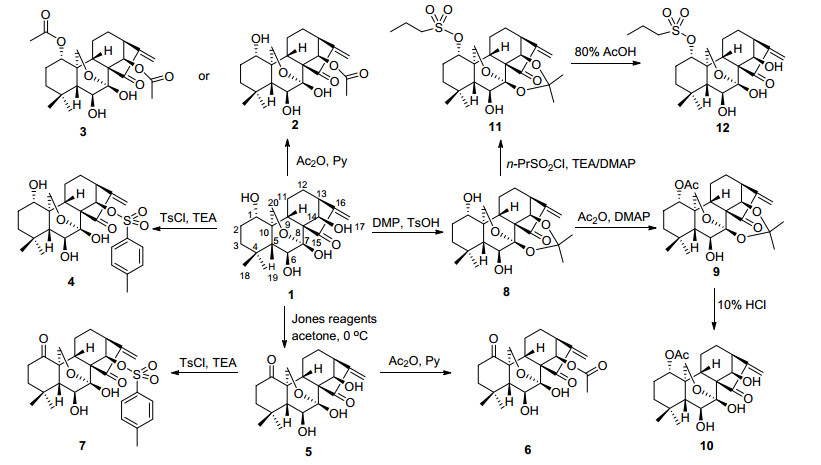
冬凌草甲素的结构修饰位点较多, 其中对C(1)-OH和C(14)-OH修饰是常采用的修饰方式之一.最简单的C(1)-OH和C(14)-OH修饰是利用这两个位点羟基活性的差异, 通过氧化或与酰氯、酸酐等一步反应进行修饰(Scheme 1), 如冬凌草甲素直接C(14) 位羟基乙酰化, 获得化合物14-乙酰冬凌草甲素(2), 通过C(1) 位、C(14) 位羟基双乙酰得1, 14-双乙酰化冬凌草甲素(3); 与对甲苯磺酰氯反应获得14-对甲苯磺酰冬凌草甲素(4); 直接以琼斯试剂氧化得1-氧代冬凌草甲素(5); 在化合物5基础上分别与乙酸酐、对甲苯磺酰氯反应可得14-乙酰-1-氧代冬凌草甲素(6)和14-对甲苯磺酰-1-氧代冬凌草甲素(7), 化合物3~7对HeLa细胞和MCF-7细胞的细胞毒毒性比冬凌草甲素强, 特别是1-氧代-冬凌草甲素[6].对冬凌草甲素C(1)-OH酰化修饰稍微复杂点, 首先冬凌草甲素通过C(7)-OH和C(14)-OH形成丙叉保护后, 对C(1)-OH乙酰化(或丙磺酰化), 再脱去C(7)、C(14) 位保护基得1-乙酰-冬凌草甲素(10)或1-丙磺酰-冬凌草甲素(12) (Scheme 1)[7], 1-乙酰-冬凌草甲素也是天然成分lasiokaurin, 具有抗原虫epimastigote活性[8].
图式1
冬凌草甲素C(1) 和C(14) 衍生物合成路线
图式1.
Synthetic route of C(1) and C(14) derivatives of oridonin
在简单修饰的基础上采取进一步修饰是普遍的选择, 通常以两种方式实现:一种是通过冬凌草甲素或其衍生物的C(14) 羟基与含羧基的1结构通过酯键相连(Scheme 2的路线A); 另一种是通过一个环状酸酐在冬凌草甲素或其衍生物的C(14) 位先引入一个羧基, 再通过新引入的羧基与含羟基或氨基的结构以酯键或酰胺键相连(Scheme 2的路线B).
 图式2
冬凌草甲素C(1) 和C(14) 衍生物合成通式
图式2.
Synthetic formula of C(1) and C(14) derivatives of oridonin
图式2
冬凌草甲素C(1) 和C(14) 衍生物合成通式
图式2.
Synthetic formula of C(1) and C(14) derivatives of oridonin
化合物13a~13i、14a~14i的制备即是通过路线A方式实现, 体外实验显示冬凌草甲素衍生物13c~13i、14c~14i对BGC-7901、SW-480、HL-60、BEL-7402、A549和B16等6种癌细胞的抑制活性强于冬凌草甲素.其中部分衍生物13h、13i、14e对H22荷瘤小鼠活性也强于冬凌草甲素和环磷酰胺.化合物13i和14e活性最强, 对HL-60细胞化合物14e的IC50值为0.84 μmol/L, 对BEL-7402细胞化合物13i的IC50值为1.00 μmol/L[8].
冬凌草甲素较高级的C(14) 位修饰则是在C(14)-OH以路线A的方式引入具有不同功能的基团或药物, 以形成杂合体, 通过协同作用发挥抗肿瘤.呋咱氮氧化物为NO供体, 通过巯基还原释放气体信使分子NO[9]. NO在生理病理方面具有多种功能, 如抗肿瘤、抗炎、血管舒张等[10].对类巨噬细胞U937, NO通过调节自噬和激活NF-κB-COX-2-IL-1β途径进一步促进冬凌草甲素诱导自噬所增强的胞葬作用[11].因此, 把冬凌草甲素和NO供体拼合形成杂合体是一种新颖的抗肿瘤候选化合物.基于这种策略, Li等以路线A方式在冬凌草甲素或其衍生物的C(14) 位引入呋咱氮氧化物, 得化合物15a~15i、16a~16i和17a~17i.所合成的冬凌草甲素呋咱氮氧化物杂合体在体外60 min都能释放较高水平的NO, 化合物15a~15i、16a~16i和17a~17i相比于冬凌草甲素(1)及1-氧代-冬凌草甲素(5), 对4种肿瘤细胞(K562、Bel-7402、MGC-803、CaEs-17) 都显示了有效的抗增殖活性, 它们的抗增殖活性与NO释放相关, NO释放越多, 抗增殖活性越强.其中化合物15h抗癌活性最强, 对K562细胞IC50值为1.82 μmol/L, 对MGC-803细胞IC50为1.81 μmol/L, 对Bel-7402细胞IC50为0.86 μmol/L[12].
基于同样的策略, Xu等[13]以路线B方式在冬凌草甲素或其衍生物的C(14) 位引入另一种NO供体(diazeni-umdiolates), 形成杂合体(18a~18d、19a~19d、20).所有的杂合体都显示了有效的抗增殖作用, IC50值在1.84~17.01 μmol/L之间, 且抗增殖作用与细胞内NO释放密切相关, 其中化合物19d活性最强, 对Bel-7402细胞其诱导凋亡能力比冬凌草甲素还强, 且随着给药剂量的增加19d能增加S期细胞的比例而降低G1期细胞的比例, 与冬凌草甲素不同, 冬凌草甲素主要阻滞细胞于G1期.可见冬凌草甲素/NO供体杂合体是有效的抗癌候选药物.
氮芥是一类强烷化剂类抗癌药物, 通过影响DNA的合成来抑制癌细胞的增殖, 但氮芥类药物选择性差, 常伴随严重的毒副作用, 易出现耐药性.把氮芥类药物与其他类药物组合是提高抗癌活性降低毒副作用的有效选择.在冬凌草甲素的C(14)-OH除了引入NO供体外, 还可以与氮芥相连. Xu等[14]以路线A方式在C(14) 位引入6种不同氮芥化合物, 合成了冬凌草甲素-氮芥杂合体(21a~21f、22a~22c、23a~23c).对K562、MCF-7、Bel-7402和MGC-803细胞, 大部分杂合体都比其母体化合物冬凌草甲素或临床应用的氮芥类药物活性强.而且代表化合物21a~21c对多药耐药细胞株SW620/ AD300和NCI-H460/MX20具有抗增殖活性.对MCF-7细胞, 化合物21b具有很强抑制活性, 其IC50值比冬凌草甲素低21倍, 且对癌细胞具有选择性.对Bel-7402细胞, 化合物21b诱导细胞凋亡, 影响其细胞周期进程.这样的合成设计对于改善氮芥的选择性具有重要意义.
氨基酸是另一类重要的物质, 具有多种生物学功能如参与蛋白质合成等生物合成途径, 与正常细胞相比在癌细胞中某些氨基酸发生了改变[15].药物通过氨基酸修饰可以提高药物水溶性、细胞渗透性、靶向性及缓释等[16].冬凌草甲素另外一种C(14)-OH修饰是通过路线B方式在冬凌草甲素的C(14) 位引入不同的氨基酸酯, 得到化合物24a~24d、25a~25f, 化合物24c、24d和25e对BGC-7901、SW-480、HL-60、BEL-7402、A549和B16等6种肿瘤细胞具有有效活性, 其中化合物24c、24d比冬凌草甲素和环磷酰胺的活性更强.因此这三个化合物具有的抗肿瘤潜力, 值得进一步深入研究[17], 也说明合成冬凌草甲素和氨基酸的杂合体是抗癌药物开发的方向之一.
针对冬凌草甲素作用机制不明确的问题, 还可以在C(14) 位引入荧光基团制备冬凌草甲素的荧光探针进行冬凌草甲素作用机制的研究.如Xu等[18]采用路线A的方式设计合成了一系列该类型化合物26a~26e、27a~27c, 其中化合物26d效果最佳, 该化合物能被肿瘤细胞快速摄取且主要定位于线粒体.通过该法证实细胞色素c在冬凌草甲素诱导的线粒体介导凋亡中起着重要作用; α, β-不饱和酮是冬凌草甲素保持活性的种重要基团, 关乎冬凌草甲素的摄取、定位及细胞毒性等.通过这种化合物设计, 对于冬凌草甲素分子作用机制的揭示具有重要的价值.
1.2 C(1)、C(2) 位[或C(1)、C(2)、C(3) 位]修饰
冬凌草甲素的C(1)、C(2) 位[或C(1)、C(2)、C(3) 位]修饰不同于单纯的C(1) 位的简单氧化或酰化修饰, 修饰的策略主要是在C(1)、C(2) 位[或C(1)、C(2)、C(3) 位]引入杂环结构或官能团等.通过这样的修饰可以提高药物的活性及改善冬凌草甲素的水溶性.对许多药物, 噻唑环是一个重要的结构片段.冬凌草甲素C(1)、C(2) 位修饰的研究是在冬凌草甲素A环C(1)、C(2) 位通过1-羰基-2-溴冬凌草甲素衍生物与硫代乙酰胺或N-取代硫脲在无保护基条件下的韩奇反应融合一个噻唑环, 这样可制得化合物33a~33h.这种修饰能把更多氮原子插入冬凌草甲素的核心骨架, 并在酸性条件下生成盐(Scheme 3).其中化合物33a~33e、33g~33h等对乳腺癌细胞, 胰腺癌细胞和前列腺癌细胞表现出有效的活性的抗增殖作用, IC50值在0.1~1 μmol/L之间, 且水溶性显著提高.这些新的化合物不仅诱导细胞凋亡, 抑制三阴-MDA-MB-231乳腺癌细胞的生长, 而且也有效对抗耐药性ER(+)MCF-7细胞[19].
 图式3
A环融合2-氨基噻唑的冬凌草甲素衍生物合成
图式3.
Synthesis of 2-amino thiazole-fused oridonin derivatives
图式3
A环融合2-氨基噻唑的冬凌草甲素衍生物合成
图式3.
Synthesis of 2-amino thiazole-fused oridonin derivatives
另外一种C(1), C(2) 位修饰则是通过一个温和且精确的方法构建了一个3, 4-二氢吡喃环插入冬凌草甲素A环的衍生物, 该反应通过反电子需求异狄尔斯阿尔德(HAD)反应获得.具体过程为通过在1-氧代-冬凌草甲素(5) A环引入环外烯酮结构, 得化合物34, 两分子化合物34通过烯酮发生自身-HAD二聚化得到二聚体化合物35, 同理也可用化合物34与乙烯基醚或二乙烯基硫发生交叉-HAD制得(Scheme 4), 该反应具有区域选择性和立体选择性, 提供了一种制备融合二氢吡喃二萜化合物的方法, 所得产物对于克服耐药性具有潜力[20].
 图式4
化合物35的合成路线
图式4.
Synthetic route of compound 35
图式4
化合物35的合成路线
图式4.
Synthetic route of compound 35
三氮唑类化合物可提高药物的靶向性及细胞穿透性, 对于C(1), C(2), C(3) 位的修饰, Ding等[21]通过Cu(Ⅰ)催化的叠氮-炔环化反应(CuAAC)在温和的条件下合成了一系列冬凌草甲素融合三氮唑的化合物.合成过程较为复杂, 首先对冬凌草甲素C(7)、C(14) 位羟基进行保护, 在此基础上在C(1) 位引入甲磺酸酯后消除获得关键中间体C(1)、C(2) 位为双键的化合物37, 以化合物37为起点分两条路线引入叠氮基, 所得的叠氮衍生物在CuI催化下再分别与苯乙炔(4-叔丁基苯乙炔)反应及5% HCl条件下脱去C(7)、C(14) 位保护基, 得到1, 2, 3-三氮唑衍生物43a、43b、45a、45b、49a、49b及相关中间体(Scheme 5).合成过程立体可控, 收率良好, 且由于在A环引入了三氮唑化合物43a、44a、45a、48b、49b对乳腺癌细胞MCF-7, MDA-MB-231抗增殖活性比冬凌草甲素强.
 图式5
A环插入三唑的冬凌草甲素衍生物合成
图式5.
Synthesis of 1, 2, 3-triazoles-installed oridonin derivatives
图式5
A环插入三唑的冬凌草甲素衍生物合成
图式5.
Synthesis of 1, 2, 3-triazoles-installed oridonin derivatives
除了在A环插入杂环之外, 还可在A环再引入一个烯酮的结构, 这样同时在冬凌草甲素的母环结构上具有了两个烯酮结构, 丰富了功能基及立体化学.可采用了两种方法引入烯酮, 一种通过消除反应在C(1)、C(2) 位引入双键, 在此基础上在C(3) 位烯丙基位氧化得到酮, 所得产物为51; 另一种则通过3, 7位重排获得一个新颖的烯酮, 得化合物58 (Scheme 6).化合物50~53、56对乳腺癌细胞MCF-7、MDA-MB-231具有显著诱导细胞凋亡抑制细胞克隆形成的作用, 相反对人正常乳腺上皮细胞与冬凌草甲素相比, 毒性相当或略低[22].
 图式6
冬凌草甲素双烯酮衍生物合成
图式6.
Synthesis of dienone oridonin derivatives
图式6
冬凌草甲素双烯酮衍生物合成
图式6.
Synthesis of dienone oridonin derivatives
2.3 C(6), C(7) 位[或C(15), C(16) 位]开环修饰
冬凌草甲素C(6)、C(7) 位连二醇通过NaIO4氧化开环反应视C(1) 位取代基的不同可得到两种类型的衍生, 当冬凌草甲素C(1) 为羟基时, 主要得延命素型衍生物59, 并可进一步通过琼斯试剂氧化得延命素型衍生物60; 当冬凌草甲素C(1)-OH氧化为羰基或乙酰化后再被NaIO4氧化开环, 则得到螺内酯型衍生物61、62 (Scheme 7).所有合成衍生物能显著抑制肿瘤细胞的增殖.细胞毒活性最强的化合物61, 对A549细胞与紫杉醇活性相当, 对Bel-7402活性稍弱于紫杉醇.对胃癌MGC-803荷瘤小鼠, 化合物61活性比其母体化合物冬凌草甲素活性还强[23].
 图式7
化合物59~62合成路线
图式7.
Synthetic route of compounds 59~62
图式7
化合物59~62合成路线
图式7.
Synthetic route of compounds 59~62
化合物59、61、62还可以进一步通过相当于冬凌草甲素的C(14)-OH与不同的含羧基结构片段相连.以化合物59为原料进行的修饰得到化合物63a~63j.对4种癌细胞(K562、MGC-803、CaEs-17、Bel-7402) 的增殖抑制活性, 所得目标化合物比冬凌草甲素及中间体化合物59都强.有些化合物如63d、63f、63h等在K562、Bel-7402、MGC-803细胞中比阳性药物紫杉醇活性还强.化合物63g对肝癌Bel-7402细胞在很低的微摩尔浓度水平就可影响细胞周期, 诱导细胞凋亡, 其作用机制研究显示是通过线粒体相关的caspase依赖性途径激发氧化应激实现[24]; 以化合物61为原料修饰得到产物(64a~64k)活性均比化合物61活性强, 与紫杉醇活性相当[23]; 以化合物62为原料修饰得到化合物65a~65j, 相比起始物62都有增强, 其中最有效的化合物65j比阳性对照紫杉醇活性还强, 对K562的IC50为0.39 μmol/L, 对肝癌Bel-7402的IC50为1.39 μmol/L.细胞内机制显示65j在低的微摩尔浓度即可诱导肝癌Bel-7402细胞凋亡.可见, 冬凌草甲素螺内酯型的衍生物对肿瘤细胞有显著的抑制活性及凋亡诱导能力[25].
另外化合物61、62可以分别和NO供体结合, 进一步在C(14) 位进行修饰制得可释放NO的不同冬凌草甲素螺内酯型衍生物66a~66f和67a~67f.所有制备的衍生物对人肿瘤细胞Bel-7402、K562、MGC-803和CaEs-17的活性均比母体化合物冬凌草甲素及化合物61、62强, 其中化合物67d抗增殖作用最强, 对4种肿瘤细胞的IC50值分别为0.86、1.74、1.16和3.75 μmol/L, 且在60 min内是所有衍生物中NO释放水平最高的衍生物, 60 min释放超过25 μmol/L.进一步的机制研究表明化合物67d在微摩尔浓度即可阻滞S期Bel-7402细胞, 通过线粒体介导途径诱导细胞凋亡[26].
冬凌草甲素的C(15)、C(16) 位开环反应则需先通过在臭氧氧化C(16)、C(17) 位双键形成C(16) 位羰基, 再在过氧化氢的条件下氧化生成C(15)、C(16) 位开环产物69, 通过化合物69上的羧基与胺反应, 得化合物70.化合物70通过两种途径最终合成了化合物72 (Scheme 8)[27].但化合物72活性未做研究, 早前报道冬凌草甲素D环上的α, β-不饱和羰基结构在衍生物中保存, 研究显示该结构片段对于冬凌草甲素的抗癌活性至关重要, 破坏该结构有损其生物活性[28], 因此这种结构修饰是否影响活性, 需进一步研究.
 图式8
化合物72合成路线
图式8.
Synthetic route of compound 72
图式8
化合物72合成路线
图式8.
Synthetic route of compound 72
2.4 C(17) 位或C(6) 位修饰
糖基化衍生物参与多种重要的生理、病理过程, 具有巨大的医药开发价值, 同时糖基化对于改善药物的水溶性、靶向性也起到重要作用[29].因此天然产物的糖基化是结构修饰的一个重要方向. Yan等[30]通过对冬凌草甲素C(7), C(14) 位保护, C(1) 位乙酰化后与保护的葡萄糖溴化物发生Koenigs-Knorr反应, 再进一步脱保护制得冬凌草甲素C(6) 位为葡萄糖基化的衍生物75, 总收率为23.0% (Scheme 9).然而该文献没有进行活性及作用机制研究.众所周知, 癌细胞需要摄取更多的葡萄糖以维持细胞的稳态、生长和增殖, 存在瓦博格效应.葡萄糖转运体GLUT1在某些肿瘤细胞中过表达[31].因此冬凌草甲素C(6) 位引入葡萄糖是否可以增加细胞摄取, 提高抗癌活性值得进一步研究.
 图式9
化合物75合成路线
图式9.
Synthetic route of compound 75
图式9
化合物75合成路线
图式9.
Synthetic route of compound 75
冬凌草甲素D环上的α, β-不饱和羰基结构片段对于冬凌草甲素的抗癌活性至关重要, 破坏该结构有损其生物活性[28].冬凌草甲素D环上的α, β-不饱和羰基结构真的不能改变吗? Yan等实验结果得出与上述矛盾的结果.他们通过在α, β-不饱和酮发生迈克尔加成反应在C(17) 位引入苯胺衍生物, 得到没有保留D环α, β-不饱和羰基结构的衍生物76a~76d (Scheme 10).这些冬凌草甲素衍生物对KB细胞的抑制活性与冬凌草甲素相当, 当R为吸电子基时活性则高于冬凌草甲素[32].
 图式10
冬凌草甲素C(16) 和C(17) 位结构修饰
图式10.
Structure modification on C(16) and C(17) positon of oridonin
图式10
冬凌草甲素C(16) 和C(17) 位结构修饰
图式10.
Structure modification on C(16) and C(17) positon of oridonin
2 冬凌草甲素结构修饰与抗分枝杆菌活性
2.1 冬凌草甲素的1位或(和)14位修饰
冬凌草甲素除了具有抗肿瘤活性外, 还具有抗分支杆菌的活性.以冬凌草甲素(1)、1-氧代-冬凌草甲素(5)和1-乙酰冬凌草甲素(10)为原料, 与不同羧酸反应, 在C(14) 位通过酯键引入不同结构片段, 所得产物77a~77d、78a~78c、79a~79l体外筛选了对Mycobacterium phlei、Mycobacterium smegmatis和Mycobacterium marinum的活性, 5个化合物(79c、79i、79j、79k、79l)对M. phlei显示了显著的抑制活性, 最小抑菌浓度(MIC)均小于2 μg/mL.结构中包含反式肉桂酸单元的化合物79k活性最强, MIC=0.5 μg/mL, 相当于抗结核药链霉素[33].化合物77d对Mycobacteriumtuberculosis H37Rv则显示了很强的活性, 其IC50值为17.1 μg/mL, 可作为先导化合物进一步研究[34].
2.2 6, 7位开环修饰
冬凌草甲素的延命素型衍生物C(14) 位修饰产物也具有抗分枝杆菌的作用.对化合物59、60分别与不同羧酸反应得化合物80a~80m及化合物81a~81k.其中化合物80c~80e、80g、80j、80l、81a~81g、81i~81k对M. phlei具有显著抑制活性, MIC值均小于2 μg/mL.其中结构中包含反式-肉桂酸片段的化合物80d、81c、81d、81j活性最强, MIC=0.5 μg/mL, 与链霉素相当[33, 34].
3 冬凌草甲素的修饰其他作用
3.1 冬凌草甲素修饰与体内过程
对冬凌草甲素C(14) 位进行修饰除了可以改善药理活性外, 还可以改善药物的体内过程如在冬凌草甲素的C(14) 位引入二硫键的衍生物(82~84), 可通过两条路线合成:一条是首先把冬凌草甲素与3-(2-吡啶二硫基)丙酸在二环己基碳二亚胺(DCC)、4-二甲氨基吡啶(DMAP)条件下反应, 所得产物82再与3-巯基-1, 2-丙二醇发生巯基-二硫键交换得终产物84; 另外一条路线为冬凌草甲素与另一个含羧基的二硫化物反应, 再脱去缩酮制得化合物84 (Scheme 11).所得产物具有生物还原响应的二硫键, 当用谷胱甘肽还原可释放出冬凌草甲素, 已通过高效液相色谱(HPLC)证明[35].但该研究没有研究其活性, 尤其是具有二硫键的化合物在谷胱甘肽(GSH)条件下可释放出硫化氢, 为H2S供体, 硫化氢为一种信使分子, 具有多种生物活性[36], 因此该课题值得在硫化氢供体的角度进一步探索.
 图式11
化合物84合成路线
图式11.
Synthetic route of compound 84
图式11
化合物84合成路线
图式11.
Synthetic route of compound 84
聚乙二醇(PEG)化广泛地应用于药物的结构修饰, 通过PEG化修饰可改善药物的水溶性及延长药物的体内循环时间[37]. Shen等[38]通过在冬凌草甲素C(14) 位与丁二酸酐反应引入一个羧基, 在此基础上与不同分子量的MeO-PEG-NH2反应, 间接在冬凌草甲素的C(14) 位进行了MeO-PEG的修饰(Scheme 12), 所得产物86水溶性提高, 其水溶性及药物释放与PEG分子量相关, PEG分子量越低, 产物水溶性越高, 高分子量的PEG修饰, 产物具有缓释作用, 而且释放具有pH响应的特点.药代动力学显示, 相比于冬凌草甲素, 产物的清除半衰期延长.可见PEG化是一个有前景的药物运输方式.对于合成的产物是否具有两亲性, 是否可自组装成纳米粒值得进一步探讨.
 图式12
化合物86合成路线
图式12.
Synthetic route of compound 86
图式12
化合物86合成路线
图式12.
Synthetic route of compound 86
3.2 转化为其他天然产物
以冬凌草甲素为起始原料合成其他天然产物是冬凌草甲素结构修饰的进一步延伸, 早期研究报道了以冬凌草甲素作为原料合成了eriocalyxin B、dehydroeriocalyxin A、14-hydroxyeriocalyxin B和14-hydroxudehydroeriocalyxin A[39]及(16R)-dihydrolongikaurin A[40].近期则报道了以冬凌草甲素为原料19步合成了semiaquilegin A (100)及其中间体87~99 (Scheme 13)[41], 可见冬凌草甲素是一个多用途的天然产物.
 图式13
Semiaquilegin A合成路线
图式13.
Synthetic route of semiaquilegin A
图式13
Semiaquilegin A合成路线
图式13.
Synthetic route of semiaquilegin A
4 前景与展望
冬凌草甲素在自然界中主要分布在香茶菜属植物, 具有抗肿瘤、抗炎和抗菌等多种生物活性, 具有潜在药用价值, 但以冬凌草甲素单体作为药物并未在临床应用, 在临床上多以其所在植物冬凌草提取物用于治疗咽炎及抗癌等.冬凌草甲素通过结构修饰可达到多种功能如提高其生物活性、改善其水溶性、改变其体内药动学过程等, 因此对冬凌草甲素的结构修饰越来越受到重视, 其结构修饰主要在C(1) 位、C(14)、C(6) 和C(7) 位等, 所获得衍生物大都表现出优良的活性.尽管冬凌草甲素的结构修饰取得了积极进展, 但也面临着一些问题: (1) 冬凌草甲素水溶性差, 但其结构修饰后有的衍生物水溶性依然没有得到改善, 对生物利用度可能具有一定影响; (2) 冬凌草甲素部分衍生物生物活性有了较大的提高, 但大都处于实验室阶段, 需要进一步药理验证走向临床; (3) 目前冬凌草甲素具体作用靶点还不十分具体, 设计的冬凌草甲素衍生作用靶点不明确.鉴于以上问题, 结合冬凌草甲素结构修饰已经取得的成就, 为今后设计出活性更佳的衍生物提供了参照.随着冬凌草甲素结构修饰的活跃, 进一步深入开展冬凌草甲素结构修饰, 尤其是设计能追踪冬凌草甲素体内过程、细胞内过程, 甚至是具体作用靶点的衍生物更具有价值, 对于打开冬凌草甲素临床应用大门具有重要意义.
-
-
[1]
Ding, Y.; Ding, C. Y.; Ye, N; Liu, Z. Q; Wold, E. A.; Chen, H. Y.; Wild, C.; Shen, Q.; Zhou, J. Eur. J. Med. Chem. 2016, 122, 102. doi: 10.1016/j.ejmech.2016.06.015
-
[2]
Marks, L. S.; Dipaola, R. S.; Nelson, P.; Chen, S.; Heber, D.; Belldegrun, A. S.; Lowe, F. C.; Fan, J.; Leaders, F. E.; Pantuck, A. J.; Tyler, V. E. Urology 2002, 60, 369. doi: 10.1016/S0090-4295(02)01913-1
-
[3]
(a) Li, C. Y.; Wang, E. Q.; Cheng, Y.; Bao, J. K. Int. J. Biochem. Cell Biol. 2011, 43, 701.
(b) Tian, W.; Chen, S. Y. Chin. J. Integr. Med. 2013, 19, 315.
(c) Liu, Z.; Ou, Y. L.; Peng, H.; Zhang, W. Z. Cell Proliferation 2012, 45, 499.
(d) Zhao, Z.; Chen, Y. Curr. Pharm. Biotechnol. 2014, 15, 1083. -
[4]
Owona, B. A.; Schluesener, H. J. Drugs R & D 2015, 15, 233.
-
[5]
Chen, J. C.; Li, W. L.; Yao, H. Q.; Xu, J. Y. Fitoterapia 2015, 103, 231. doi: 10.1016/j.fitote.2015.04.012
-
[6]
伍旭, 赵烽, 刘珂, 中草药, 2009, 40, 348. doi: 10.3321/j.issn:0253-2670.2009.03.004Wu, X.; Zhao, F.; Liu, K. Chin. Tradit. Herbal. Drugs 2009, 40, 348(in Chinese). doi: 10.3321/j.issn:0253-2670.2009.03.004
-
[7]
闫学斌, 李雯, 刘宏民, 郑州大学学报(工学版), 2006, 27, 113. doi: 10.3969/j.issn.1671-6833.2006.02.029Yan, X. B.; Li, W.; Liu, H. M. J. Zhengzhou Univ. (Eng. Sci.) 2006, 27, 113(in Chinese). doi: 10.3969/j.issn.1671-6833.2006.02.029
-
[8]
Xu, J. Y.; Yang, J. Y.; Ran, Q.; Wang, L.; Liu, J.; Wang, Z. X.; Wu, X. M.; Hua, W, Y.; Yuan, S. T.; Zhang, L. Y.; Shen, M. Q.; Ding, Y. F. Bioorg. Med. Chem. Lett. 2008, 18, 4741. doi: 10.1016/j.bmcl.2008.06.097
-
[9]
Ferioli, R.; Folco, G. C.; Ferretti, C.; Gasco, A. M.; Medana, C.; Fruttero, R.; Civelli, M.; Gasco, A. Br. J. Pharmacol. 1995, 114, 816. doi: 10.1111/bph.1995.114.issue-4
-
[10]
Carpenter, A. W.; Schoenfisch, M. H. Chem. Soc. Rev. 2012, 41, 3742. doi: 10.1039/c2cs15273h
-
[11]
Zhang, L.; He, H.; Ye, Y.; Liu, W.; Fan, S.; Tashiro, S.; Onodera, S.; Ikejima, T. Free Radical Res. 2012, 46, 1207. doi: 10.3109/10715762.2012.700515
-
[12]
(a) Li, D. H.; Wang, L.; Cai, H.; Jiang, B. W.; Zhang, Y. H.; Sun, Y. J.; Xu, J. Y. Chin. J. Nat. Med. 2012, 10, 471.
(b) Li, D. H.; Wang, L.; Cai, H.; Zhang, Y. H.; Xu, J. Y. Molecules 2012, 17, 7556. -
[13]
Xu, S. T.; Wang, G. Y.; Lin, Y.; Zhang, Y. J.; Pei, L. L.; Yao, H.; Hu, M.; Qiu, Y. Y.; Huang, Z. J.; Zhang, Y. H.; Xu, J. Y. Bioorg. Med. Chem. Lett. 2016, 26, 2795. doi: 10.1016/j.bmcl.2016.04.068
-
[14]
Xu, S. T.; Pei, L. L.; Wang, C. Q.; Zhang, Y. K.; Li, D. H.; Yao, H. Q.; Wu, X. M.; Chen, Z. S.; Sun, Y. J.; Xu, J. Y. ACS Med. Chem. Lett. 2014, 5, 797. doi: 10.1021/ml500141f
-
[15]
Tsun, Z. Y.; Possemato, R. S. Cell Dev. Biol. 2015, 43, 22. doi: 10.1016/j.semcdb.2015.08.002
-
[16]
Vig, B. S.; Huttunen, K. M.; Laine, K.; Rautio, J. Adv. Drug Delivery Rev. 2013, 65, 1370. doi: 10.1016/j.addr.2012.10.001
-
[17]
Wang, L.; Ran, Q.; Li, D. H.; Yao, H. Q.; Zhang, Y. H.; Yuan, S. T.; Zhang, L. Y.; Shen, M. Q.; Xu, J. Y. Chin. J. Nat. Med. 2011, 9, 194.
-
[18]
Xu, S. T.; Luo, S. S.; Yao, H.; Cai, H.; Miao, X. M.; Wu, F.; Yang, D. H.; Wu, X. M.; Xie, W. J.; Yao, H. Q.; Chen, Z. S.; Xu, J. Y. J. Med. Chem. 2016, 59, 5022. doi: 10.1021/acs.jmedchem.6b00408
-
[19]
Ding, C. Y.; Zhang, Y. S.; Chen, H. J.; Yang, Z. D.; Wild, C.; Chu, L. L.; Liu, H. L.; Shen, Q.; Zhou, J. J. Med. Chem. 2013, 56, 5048. doi: 10.1021/jm400367n
-
[20]
Ding, C. Y.; Wang, L. L.; Chen, H. J.; Wild, C.; Ye, N.; Ding, Y.; Wang, T. Z.; White, M. A.; Shen, Q.; Zhou, J. Org. Biomol. Chem. 2014, 12, 8442. doi: 10.1039/C4OB01040J
-
[21]
Ding, C. Y.; Zhang, Y. S.; Chen, H. J.; Wild, C.; Wang, T. Z.; White, M. A.; Shen, Q.; Zhou, J. Org. Lett. 2013, 15, 3718. doi: 10.1021/ol4015865
-
[22]
Ding, C. Y.; Zhang, Y. S.; Chen, H. J.; Yang, Z. D.; Wild, C.; Ye, N.; Ester, C. D.; Xiong, A.; White, M. A.; Shen, Q.; Zhou, J. J. Med. Chem. 2013, 56, 8814. doi: 10.1021/jm401248x
-
[23]
Wang, L.; Li, D. H.; Xu, S. T.; Cai, H.; Yao, H. Q.; Zhang, Y. H.; Jiang, J. Y.; Xu, J. Y. Eur. J.Med. Chem. 2012, 52, 242. doi: 10.1016/j.ejmech.2012.03.024
-
[24]
Li, D. H.; Xu, S. T.; Cai, H.; Pei, L. L.; Zhang, H. Y.; Wang, L.; Yao, H. Q.; Wu, X. M.; Jiang, J. J.; Sun, Y. J.; Xu, J. Y. Eur. J. Med. Chem. 2013, 64, 215. doi: 10.1016/j.ejmech.2013.04.012
-
[25]
Li, D. H.; Cai, H.; Jiang, B. W.; Liu, G. Y.; Wang, Y. T.; Wang, L.; Yao, H. Q.; Wu, X. M.; Sun, Y. J.; Xu, J. Y. Eur. J. Med. Chem. 2013, 59, 322. doi: 10.1016/j.ejmech.2012.11.002
-
[26]
Li, D. H.; Han, T.; Tian, K. T.; Tang, S.; Xu, S. T.; Hu, X.; Wang, L.; Li, Z. L.; Hua, H. M.; Xu, J. Y. Bioorg. Med. Chem. Lett. 2016, 26, 4191. doi: 10.1016/j.bmcl.2016.07.059
-
[27]
Zhang, M.; Zhang, Y. M.; Lu, W.; Nan, F. J. Org. Biomol. Chem. 2011, 9, 4436. doi: 10.1039/c1ob05611e
-
[28]
Huang, S. X.; Zhou, Y.; Pu, J. X.; Lia, R. T.; Lia, X.; Xiao, W. L.; Lou, L. G.; Han, Q. B.; Ding, L. S.; Peng, S. L.; Sun, H. D. Tetrahedron 2006, 62, 4941. doi: 10.1016/j.tet.2006.02.079
-
[29]
Dalziel, M.; Crispin, M.; Scanlan, C. N.; Zitzmann, N.; Dwek, R. A. Science 2014, 343, 1235681. doi: 10.1126/science.1235681
-
[30]
闫学斌, 雷萌, 张建业, 刘宏民, 有机化学, 2005, 25, 222. doi: 10.3321/j.issn:0253-2786.2005.02.017Yan, X. B.; Lei, M.; Zhang, J. Y.; Liu, H. M. Chin. J. Org. Chem. 2005, 25, 222(in Chinese). doi: 10.3321/j.issn:0253-2786.2005.02.017
-
[31]
Szablewski, L. Biochim. Biophys. Acta 2013, 1835, 164.
-
[32]
闫学斌, 张建业, 可钰, 周霞, 刘宏民, 郑州大学学报(医学版), 2007, 42, 39. doi: 10.3969/j.issn.1671-6825.2007.01.015Yan, X. B.; Zhang, J. Y.; Yu, K.; Zhou, X.; Liu, H. M. J. Zhengzhou. Univ. (Med. Sci.) 2007, 42, 39(in Chinese). doi: 10.3969/j.issn.1671-6825.2007.01.015
-
[33]
Xu, S. T.; Li, D. H.; Pei, L. L.; Yao, H.; Wang, C.; Cai, H. Q.; Yao, H.; Wu, X. M.; Xu, J. Y. Bioorg. Med. Chem. Lett. 2014, 24, 2811. doi: 10.1016/j.bmcl.2014.04.119
-
[34]
Xu, S. T.; Pei, L. L.; Li, D. H.; Yao, H.; Cai, H.; Yao, H. Q.; Wu, X. M.; Xu, J. Y. Fitoterapia 2014, 99, 300. doi: 10.1016/j.fitote.2014.10.005
-
[35]
Du, M.; Guo, Q. S.; Feng, H.; Lu, G. L.; Huang, X. Y. Chin. J. Chem. 2014, 32, 448. doi: 10.1002/cjoc.201300761
-
[36]
Song, Z. J.; Ying, M.; Lee, Z. W.; Dai, W. L.; Hagen, T.; Moore, P. K.; Huang, D. J.; Deng, L. W.; Tan, C. H. Med. Chem. Commun. 2014, 5, 557. doi: 10.1039/C3MD00362K
-
[37]
Veronese, F.; Mero, A. Biodrugs 2008, 22, 315. doi: 10.2165/00063030-200822050-00004
-
[38]
Shen, J. Y.; Zhang, D. R.; Zhao, Z. X.; Jia, L. J.; Zheng, D. D.; Liu, G. P.; Hao, L. L.; Zhang, Q. Q.; Tian, X. N.; Li, C. Y.; Guo, H. J. Int. J. Pharm. 2013, 456, 80. doi: 10.1016/j.ijpharm.2013.08.014
-
[39]
周维善, 程云行, 化学学报, 1990, 48, 1185.Zhou, W. S.; Chen, Y. X. Acta Chim. Sinica 1990, 48, 1185(in Chinese).
-
[40]
Takeda, Y.; Fujita, T. Chem. Inform. 1988, 2, 379.
-
[41]
Li, J.; Jiang, Y.; Li, Q. J.; Jia, Y. X.; Tu, P. F. Tetrahedron Lett. 2010, 51, 1121. doi: 10.1016/j.tetlet.2009.12.118
-
[1]
-

图式1 冬凌草甲素C(1) 和C(14) 衍生物合成路线
Scheme 1 Synthetic route of C(1) and C(14) derivatives of oridonin

图式2 冬凌草甲素C(1) 和C(14) 衍生物合成通式
Scheme 2 Synthetic formula of C(1) and C(14) derivatives of oridonin

图式3 A环融合2-氨基噻唑的冬凌草甲素衍生物合成
Scheme 3 Synthesis of 2-amino thiazole-fused oridonin derivatives

图式5 A环插入三唑的冬凌草甲素衍生物合成
Scheme 5 Synthesis of 1, 2, 3-triazoles-installed oridonin derivatives

图式10 冬凌草甲素C(16) 和C(17) 位结构修饰
Scheme 10 Structure modification on C(16) and C(17) positon of oridonin
-


 下载:
下载:












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 39
- 文章访问数: 5559
- HTML全文浏览量: 1494
 下载:
下载:
