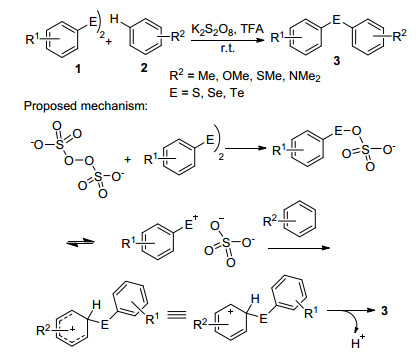
 图式1
K2S2O8催化富电子苯的C—H硫/硒芳基化反应
图式1.
K2S2O8-catalyzed C—H sulfenylation/selenation of electron rich benzenes using disulfides/diselenides
图式1
K2S2O8催化富电子苯的C—H硫/硒芳基化反应
图式1.
K2S2O8-catalyzed C—H sulfenylation/selenation of electron rich benzenes using disulfides/diselenides
引用本文:
刘云云, 熊进, 韦丽. 无过渡金属催化的C(sp2)-H键官能化反应构建C(sp2)-S键研究进展[J]. 有机化学,
2017, 37(7): 1667-1680.
doi:
10.6023/cjoc201702009

Citation: Liu Yunyun, Xiong Jin, Wei Li. Recent Advances in the C(sp2)-S Bond Formation Reactions by Transition Metal-Free C(sp2)-H Functionalization[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1667-1680. doi: 10.6023/cjoc201702009
Citation: Liu Yunyun, Xiong Jin, Wei Li. Recent Advances in the C(sp2)-S Bond Formation Reactions by Transition Metal-Free C(sp2)-H Functionalization[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1667-1680. doi: 10.6023/cjoc201702009
无过渡金属催化的C(sp2)-H键官能化反应构建C(sp2)-S键研究进展
摘要:
含有C(sp2)-S键的芳基/烯基硫醚类化合物广泛存在于具有生物活性或其它潜在应用价值的有机化合物中.在过去的十来年时间,构建C(sp2)-S键的相关研究发展迅猛,大量新型高效的催化体系和丰富多样的反应底物被开发报道,极大地丰富了含有C(sp2)-S骨架的化合物合成的研究方法学.综述了2001~2016年期间通过无过渡金属催化的碳-氢键官能化反应构建C(sp2)-S键的研究进展.
-
关键词:
- 无过渡金属
- / C (sp2)-H键
- / 芳(杂)环
- / 烯基
- / C(sp2)-S键构建
English
Recent Advances in the C(sp2)-S Bond Formation Reactions by Transition Metal-Free C(sp2)-H Functionalization
Abstract:
Aryl/vinyl thioethers containing C(sp2)-S bonds are prevalent in biologically relevant molecules and compounds with other potential applications. Around the past decade, rapid progress has taken place in the research area of C(sp2)-S construction, and a large number of efficient new catalytic systems as well as structurally versatile substrates have been identified for such reactions, which significantly enriched the content of the synthetic methodologies on C(sp2)-S bond formation. This review introduces mainly the research advances on transition metal-free C(sp2)-S bond forming reaction by means of C(sp2)-H bond functionalization over the period of 2001~2016.
-
Key words:
- transition metal-free
- / C(sp2)-H bond
- / (Hetero)aryl
- / vinyl
- / C(sp2)-S bond formation
-
含碳-硫键的有机化合物如硫醚类广泛存在于天然产物、药物和材料分子中, 在许多领域都有着广泛而重要的应用价值[1].此外, 许多其它含硫化合物在治疗人类免疫缺陷病毒(HIV)、癌症、阿尔茨海默症及帕金森症等方面展示了良好的应用前景[2].因此, 寻找简单高效的方法来构建C—S键引起了有机化学工作者研究兴趣.其中, 过渡金属催化下卤代化合物或硼酸类化合物和硫醇或二硫醚等含硫试剂的偶联反应、一些亲核取代反应如Mitsunobu反应以及不饱和键的加成反应等是构建C—S键的传统方法[3].由于这些合成方法通常需要在高温条件下使用过渡金属作为催化剂或需要酸、碱、氧化添加物等, 在原子经济性和环境友好性等方面存在限制.近几年来, 随着对的直接C—H官能团化反应研究的深入, 该方法也成为构建C—S键的重要手段.在已知基于C—H键转化直接构建C—S键的方法中, 其中一类是过渡金属催化下的C—H活化反应, 这类合成方法在底物适用性和反应条件的温和性方面具有显著优势, 然而过渡金属特别是贵金属催化剂的使用仍然是需要解决的问题, 尤其在药物或药物中间体的合成中, 金属的使用因为残留问题而难以有效应用[4].相比而言, 另外一类合成方法, 即无金属催化条件下的C—H官能化反应构建C—S键则在原子经济性和合成洁净性方面具有独特优势.最近十多年来, 大量经济、高效的基于C—H键官能化的C—S键构建方法被陆续报道.由于含C—S键化合物的重要性以及相关合成研究的快速发展, 本文对近期基于无过渡金属条件下C—H官能化反应构建C(sp2)—S键的相关研究进展进行综述.
1 芳基C(sp2)—H键硫醚化反应
1.1 非杂环芳香化合物的C(sp2)—H键硫醚化反应
对于非杂环芳香化合物, 通常是含有各种给电子基团的富电子芳香化合物可以进行C—H键的直接官能化反应. 2013年Kumar研究小组[5]报道了二芳基二硫醚(1)和含推电子取代基如烷基、烷氧基、硫烷基以及氨基等的苯衍生物2之间的硫芳基化反应, 反应以过硫酸钾为氧化剂, 三氟乙酸为溶剂, 室温即可进行, 提供了一种温和高效地合成二芳基硫醚的方法.同时, 在该反应条件下, 二芳基二硒/碲醚类化合物同样具有很好的适用性, 可用于合成相应二芳基硒醚和碲醚.作者推测该反应的机理是亲电取代反应, 即二芳基二硫/硒/碲醚类化合物和过硫酸根作用得到活性的芳基硫/硒/碲阳离子前体, 该阳离子和富电子芳烃经历亲C—H键的亲电取代反应得到目标产物3 (Scheme 1).
图式1
K2S2O8催化富电子苯的C—H硫/硒芳基化反应
图式1.
K2S2O8-catalyzed C—H sulfenylation/selenation of electron rich benzenes using disulfides/diselenides
2014年, Fu等[6]以BF3•OEt2为催化剂, 在不需要惰性气体保护以及其它额外添加物条件下, 报道了以N-硫代丁二酰亚胺4和苯酚类化合物5的硫芳基化反应, 在室温条件下实现了含苯酚片段的硫醚类化合物6合成.该方法采用硫代丁二酰亚胺为硫芳基化试剂, 对含有吸电子或给电子取代基的苯酚都展示了良好的适用性, 以较好的产率得到了不同的双芳基硫醚产物(A, Scheme 2).随后, Cossy小组[7]同样以N-硫代丁二酰亚胺为硫化试剂, 成功实现了富电子芳烃类化合物的硫化反应(B, Scheme 2).该反应室温条件下即可顺利进行, 通过相应的硫化试剂, 不仅可以实现双芳基硫醚类化合物的合成, 而且可以实现芳基烷基硫醚的合成.与之前的合成方法相比, 该方法具有更好的底物适用范围和区域选择性.不足之处是需要用到大大过量的三氟乙酸(TFA)作为添加物.
 图式2
N-硫代丁二酰亚胺为硫源的富电子苯C—H硫芳基化反应
图式2.
N-Thiobutanimides as thio sources for the sulfenylation of electron rich benzenes
图式2
N-硫代丁二酰亚胺为硫源的富电子苯C—H硫芳基化反应
图式2.
N-Thiobutanimides as thio sources for the sulfenylation of electron rich benzenes
2015年, Wang课题组[8]报道了以二叔丁基过氧化物(DTBP)为氧化剂, 氮气保护条件下碘催化的苯胺类化合物和硫酚之间的直接偶联反应, 建立了通过苯胺衍生物的芳基C—H键转化直接合成相应二芳基硫醚的方法(A, Scheme 3).研究发现, 苯胺及取代苯胺发生硫醚化反应时, 由于氨基位阻的影响, 通常在氨基的对位进行.而当氨基的对位被其它原子或基团取代后, 硫醚化反应可发生在邻位.此后, Yang等[9]采用类似反应条件研究了连、偏、均三甲氧基苯及富电子苯并芳杂环类化合物13和硫酚/硫醇12的硫醚化反应.研究发现具有多个不同反应位点的连、偏三甲氧基苯在该反应条件下只得到一个硫醚化产物(B, Scheme 3).
 图式3
硫酚/硫醇为硫源的富电子苯C—H硫芳基化反应
图式3.
Thiophenols/thiols as thio sources for the sulfenylation of electron rich benzenes
图式3
硫酚/硫醇为硫源的富电子苯C—H硫芳基化反应
图式3.
Thiophenols/thiols as thio sources for the sulfenylation of electron rich benzenes
Peddinti等[10]以I2/DMSO催化体系进行富电子芳香化合物15, 包括苯、萘衍生物和硫酚之间的偶联反应, 顺利实现了双硫醚类化合物16的合成.此外, 作者对底物进行扩展研究时, 发现该反应体系中同样可以顺利进行香豆素类化合物17和二硫缩烯酮19中C(sp2)—H键的硫芳基化反应, 以优秀的产率分别得到硫醚类产物18和20.根据作者推测, 反应由碘和硫酚结合生成芳基硫碘活性中间体引发.芳基硫碘形成后原位和活化的C(sp2)—H键供体如萘酚先发生加成反应, 然后消除HI得到硫醚类产物. HI在二甲亚砜的氧化作用下可以再次生成单质碘并参与循环催化反应(Scheme 4).
 图式4
碘催化富电子苯和极性烯基C—H硫芳基化反应
图式4.
I2-catalyzed C—H sulfenylation of electron rich benzenes polor vinyls
图式4
碘催化富电子苯和极性烯基C—H硫芳基化反应
图式4.
I2-catalyzed C—H sulfenylation of electron rich benzenes polor vinyls
Baumgartner等[11]曾在研究硝基烷烃类化合物的硫芳基化反应中发现二芳基二硫醚作为硫源和萘酚发生反应可得到相应的羟基萘硫醚, 反应需通过分别采用叔丁醇钾和盐酸分两步处理完成. Prabhu小组[12]通过苯并噁唑-2-硫酮/苯并噻唑-2-硫酮21在三氟甲磺酸(TfOH)催化下异构化为相应的硫醇, 以过硫酸钾为氧化剂, 硫醇作为硫源和苯酚/苯甲醚类化合物22反应, 在60 ℃加热可发生脱氢偶联反应得到硫醚类产物23.当采用富电子杂环类化合物如噻吩/苯并噻吩24作为另一反应底物时, 在同样氧化剂和酸催化作用下, 可得到相应的双杂环硫醚产物25, 反应在室温即可进行(Scheme 5).
 图式5
K2S2O8/TfOH促进的苯基醚和吲哚C—H硫芳基化反应
图式5.
K2S2O8/TfOH-promoted C—H sulfenylation of phenyl ethers and indoles
图式5
K2S2O8/TfOH促进的苯基醚和吲哚C—H硫芳基化反应
图式5.
K2S2O8/TfOH-promoted C—H sulfenylation of phenyl ethers and indoles
Huang等[13]以磺酰肼类化合物28为硫化试剂, 在碘为催化剂, 100 ℃加热条件下, 实现了了2-萘酚26/2-萘胺27的硫芳基化反应, 在反应条件下, 1-位的碳-氢键选择性发生的硫醚化反应(A, Scheme 6).当采用相应的1-萘酚/1-萘胺进行反应时, 碳-氢硫醚化反应在4-位进行.随后, Lu课题组[14]将磺酰肼为硫源的芳基C—H键硫醚化反应的底物范围扩展到普通的非富电子基团活化苯衍生物, 反应同样以碘为催化剂, 在120 ℃条件下, 以1, 2-二氯乙烷(DCE)为溶剂, 合成了系列芳基硫下, 以1, 2-二氯乙烷(DCE)为溶剂, 合成了系列芳基硫醚类化合物.除了传统的苯/萘酚及醚等底物, 甲基苯、甲基苯硫醚以及卤代苯都可发生反应, 不足之处在于发生官能化反应的碳-氢键位置主要取决于底物结构(B, Scheme 6).
 图式6
磺酰肼为硫源的富电子苯/萘的C—H硫醚化反应
图式6.
Sulfonyl hydrazine as thiol sources in the C—H thiolation of electron benzenes/naphthalenes
图式6
磺酰肼为硫源的富电子苯/萘的C—H硫醚化反应
图式6.
Sulfonyl hydrazine as thiol sources in the C—H thiolation of electron benzenes/naphthalenes
Lin课题组[15]以I2/PhPh3催化体系, 水为绿色溶剂, 在空气氛围下顺利实现了苯亚磺酸钠33和富电子芳香化合物的硫醚化反应, 通过C(sp2)—H功能化, 完成了一系列硫醚化合物的合成, 并且在这一催化条件下将反应放大到克级也能得到较高产率(A, Scheme 7).随后, 该课题组[16]再次实现了以廉价易得的苯磺酰氯34为硫源的芳香化合物硫醚化反应.反应以TBAI-HBr为催化剂, PEG400作为溶剂并在100 ℃加热.该反应条件不仅适合富电子的酚/芳醚类底物, 同时吲哚以及吡唑酮类化合物的也能顺利的进行C—H硫醚化反应, 为合成富电子芳基硫醚类化合物提供一条新的无金属催化途径(B, Scheme 7).
 图式7
亚磺酸钠和磺酰氯为硫源的富电子苯/萘的C—H硫醚化反应
图式7.
Sodium sulfinates and sufonyl chlorides as thiol sources in the C—H thiolation of electron benzenes/naphthalenes
图式7
亚磺酸钠和磺酰氯为硫源的富电子苯/萘的C—H硫醚化反应
图式7.
Sodium sulfinates and sufonyl chlorides as thiol sources in the C—H thiolation of electron benzenes/naphthalenes
最近, Zhao和Lu等[17]报道了黄酮衍生物35和磺酰肼28之间的偶联反应.反应仅需要使用单质碘为催化剂, 在乙醇介质中可实现黄酮苯环上C—H键的硫芳基/烷基化反应得到含硫醚结构的黄酮类化合物36.该反应操作简单, 而且通过底物结构上预留的官能团, 可以很好地控制反应的区域选择性(Eq. 1).
1.2 杂环芳香化合物的C—H键硫醚化反应
在芳杂环化合物中, 可在无过渡金属催化条件下实现C—H键硫醚化的最典型化合物为吲哚. Kita等[18]早期以O, S-缩醌酮(quinine O, S-acetals) 37作为硫源和吲哚38进行反应, 在三氟甲磺酸三甲基硅酯催化下, 可以得到3-位硫醚化的吲哚39.同时, 该催化方法也适用于烯醇硅醚40和富电子苯衍生物42的C—H键硫醚化反应, 分别得到硫醚产物41和43 (Scheme 8).利用该类硫源进行C—H键硫化反应合成硫醚的方法随后被扩展到吡咯、噻吩等其它常见的富电子芳杂环化合物[19].该反应方法的限制主要在于需要预先合成硫源37, 故而操作不够简便.
 图式8
O, S-缩醌酮为硫源的吲哚和富电子烯醚/苯基醚的C—H硫醚化反应
图式8.
Quinine O, S-acetals as thiol sources for the C—H thioetherification of indoles, electron rich vinyl/phenyl ethers
图式8
O, S-缩醌酮为硫源的吲哚和富电子烯醚/苯基醚的C—H硫醚化反应
图式8.
Quinine O, S-acetals as thiol sources for the C—H thioetherification of indoles, electron rich vinyl/phenyl ethers
采用更加常见和易得的硫源进行包括吲哚在内的芳杂环化合物C—H键硫醚化反应在近几年取得了较大进展, 2004年, Schlosser小组[20]报道了N-氯代丁二酰亚胺(NCS)促进下2-取代的吲哚和硫酚/硫醇反应合成吲哚硫醚的方法.反应以二氯甲烷为溶剂, 首先需要在-78 ℃条件下进行投料, 随后升温到0 ℃后反应15 min, 产率中等到优秀(Eq. 2). Yadav等[21]以商业易得的氟试剂SelectfluorTM为氧化剂, 同样实现了硫酚/硫醇和吲哚的C-3硫醚化反应, 在该反应条件下吲哚的1-和2-号位均不需要保护, 同时反应条件比较温和, 室温条件下可以较高产率得到一系列吲哚硫醚类化合物. Liu等[22]发现采用硫酚类化合物作为硫源, 仅以NaOH为碱性促进剂也可以高效实现吲哚3-位碳C—H的硫醚化反应, 反应在70 ℃加热条件下就能顺利进行.最近, Huang等[23]报道了以碘为催化剂, H2O2氧化剂, 水为绿色溶剂条件下吲哚和硫酚/硫醇类化合物的C—H偶联合成吲哚硫醚的反应, 反应操作简单, 环境友好, 不仅适用于苯硫酚类化合物, 同时对于芳杂硫酚类化合物同样适用, 且产率良好.随后, 采用血清蛋白-碘共催化的方法在水相中也成功实现了这类反应[24].
Li等[25]直接NaOH为催化剂, 异丙醇和水为混合溶剂、在120 ℃加热条件下实现了吲哚和巯基苯甲酸44之间的C—H键硫醚化, 合成了一系列含苯甲酸次级结构的吲哚硫醚类化合物45 (Eq. 3).随后他们研究小组[26]将上述反应体系扩展到邻巯基苯甲酸的二硫醚类化合物和吲哚的C—H硫醚化反应, 在130 ℃加热条件下实现了类似产物的合成.
硫酚/硫醇作为最常见的硫源, 用于进行合成反应时避免了额外的原料合成步骤, 但是绝大部分硫酚/硫醇本身都具有的刺激性气味, 对于相关的合成操作产生了较大的不便.因此, 发展简单易得、结构稳定的硫源进行C—H键的硫醚化反应也是化学工作者关注的重要研究内容. Wei等[27]在2012年报道了以二芳基二硫醚类化合物46为硫源合成吲哚-3-硫醚类化合物39的反应.反应以碘为催化剂, DMSO为氧化剂以及环境友好的碳酸二甲酯(DMC)为绿色溶剂, 在40 ℃加热条件下可顺利进行, 该体系对二芳基二硫醚以及二烷基二硫醚都具有适用性(Scheme 9).同年, Chen和Wu等[28]采用NBS为促进剂, 在DMF, -15或-10 ℃条件下实现了类似的合成反应, 不过反应仅对二芳基二硫醚类底物适用(Scheme 9).
 图式9
二硫醚为硫源的吲哚3-C—H硫醚化反应
图式9.
Disulfides as thio sources for the 3-C—H thioetherification of indoles
图式9
二硫醚为硫源的吲哚3-C—H硫醚化反应
图式9.
Disulfides as thio sources for the 3-C—H thioetherification of indoles
随后, Zhang等[29]在空气氛围下, 以DMSO为溶剂, 仅使用催化量的碳酸钾并将反应加热到100 ℃同样实现了这一基于二硫醚硫源的吲哚硫醚化反应.在这一简化反应条件下, 2-取代吲哚及非取代吲哚都可以顺利进行反应, 以优秀的产率得到目标产物.同时, 将合成反应放大到克级也具有良好的反应效果(Scheme 10).
 图式10
K2CO3催化下二硫醚和吲哚的3-C—H硫芳基化反应
图式10.
K2CO3-catalyzed indole 3-C—H sulfenylation using disulfides
图式10
K2CO3催化下二硫醚和吲哚的3-C—H硫芳基化反应
图式10.
K2CO3-catalyzed indole 3-C—H sulfenylation using disulfides
Kumar等[30]利用过硫酸铵为氧化剂, 甲醇为溶剂, 加热回流条件下, 以芳基二硫醚为硫源顺利实现了吲哚的C-3位的硫醚化反应, 同时在该反应条件下芳基二硒醚也可以顺利地和吲哚在C-3位发生硒化反应.值得一提的是, 当在反应体系中额外加入分子碘时, 可以同时实现吲哚C-2和C-3硫醚化反应, 得到吲哚双硫醚产物47 (Scheme 11). Braga小组[31]以碘为催化剂, DMSO为氧化剂, 实现了微波促进下的二硫醚类化合物和吲哚的C-3硫醚化反应, 同时在该反应条件下, 二硒醚类化合物也可以和吲哚发生反应, 高产率的生成相应的C-3硒化吲哚衍生物.
 图式11
过硫酸铵促进二硫醚为硫源的吲哚3-以及2, 3-二硫芳基化反应
图式11.
(NH4)2S2O8-promoted 3-and 2, 3-(di)sulfenylation of indoles using disulfides
图式11
过硫酸铵促进二硫醚为硫源的吲哚3-以及2, 3-二硫芳基化反应
图式11.
(NH4)2S2O8-promoted 3-and 2, 3-(di)sulfenylation of indoles using disulfides
Deng小组[32]研究发现在碘为催化剂, DMSO为氧化剂, 二乙基磷酸酯为添加物以及100 ℃加热条件下, 亚磺酸钠48可以作为硫源和吲哚进行C—H键硫醚化反应, 生成吲哚硫醚产物.该体系中芳基/烷基亚磺酸钠作为底物都能有效得到目标化合物(Scheme 12).随后, Kuhakarn等[33]使用相对更加简单的I2/PhPh3催化体系, 在乙醇为溶剂80 ℃的加热条件下, 也顺利实现了这一反应(Scheme 12). Rao等[34]在同年发展了以过硫酸钾(K2S2O8)作为氧化剂, 在加热条件下产生硫酸根自由基引发反应的方法, 实现了芳基亚磺酸钠作为硫源和吲哚进行C—H硫化反应合成吲哚硫醚39的转化.
 图式12
亚磺酸钠为硫源的吲哚3-C—H硫醚化反应
图式12.
3-C—H thioetherification of indoles using sodium sulfinates as thio sources
图式12
亚磺酸钠为硫源的吲哚3-C—H硫醚化反应
图式12.
3-C—H thioetherification of indoles using sodium sulfinates as thio sources
和亚磺酸钠一样, 磺酰氯类化合物在有机合成中也常用做硫源参加C—S键的构建反应. Kumaraswamy等[35]报道了以磺酰氯为硫源和吲哚反应合成吲哚硫醚的方法.该反应以碘单质为催化剂, 空气氛围下, 不需要额外添加任何自由基引发剂, 在1, 4-二氧六环中加热至80 ℃顺利实现了多种含N-取代或N-非取代结构吲哚硫醚类化合物的高效合成(Eq. 4). Zhao和Lu等[36]最近则报道了PPh3/KI在乙醇介质、100 ℃加热的催化方法实现同样的反应, 而且该催化方法可以扩展到富电子的吡唑酮, 合成羟基吡唑硫醚类化合物.
2013年, Tian等[37]首次以磺酰肼类化合物为硫源, 实现了无金属催化条件下吲哚的C-3硫醚化反应.这一偶联反应在碘催化剂存在下, 乙醇中加热到70 ℃即可高效进行, 反应操作简单, 条件温和, 且不需要惰性气体保护.同时, 该方法以磺酰肼类化合物为硫源, 避免了使用具有刺激性气味的硫酚或硫醇类化合物.同时研究还发现在催化条件下, 吲哚C-3位优先发生硫醚化反应, 而当3-位被占据时, 反应则发生在C-2位, 得到产物为吲哚-2-硫醚51.反应的可能机理是磺酰肼在碘的氧化作用下经历脱除次碘酸以及氮气的过程得到活性硫碘中间体, 该中间体和吲哚反应得到吲哚硫醚类产物, 同时释放出HI.次碘酸和HI之间的氧化还原反应重新生成分子点进行催化(Scheme 13).最近, Wang等[38]研究发现, 在以水作为介质, 加热至140 ℃时, 不需要催化剂也可以实现同样的合成吲哚-3-硫醚的反应. Barman等[39]则发展了以基于1, 8-二氮杂二环十一碳-7-烯(DBU)的离子液为介质, 微波辐射促进的方法, 在100 ℃下实现了这一反应.
 图式13
磺酰肼为硫源的吲哚3-和2-C—H硫醚化反应
图式13.
3-and 2-C—H Thioetherification of indoles using sulfonyl hydrazines as thio sources
图式13
磺酰肼为硫源的吲哚3-和2-C—H硫醚化反应
图式13.
3-and 2-C—H Thioetherification of indoles using sulfonyl hydrazines as thio sources
通过采用季铵盐四丁基碘化铵(n-Bu4NI)和对甲苯磺酸为共催化体系以及二氯乙烷为溶剂, Liu等[40]报道了亚磺酸52和吲哚的C—H键硫醚化反应, 以良好到优秀的产率实现了吲哚-3-硫醚类化合物的合成, 芳基及烷基亚硫酸在催化反应中都有很好的适用性(Eq. 5). Barman等[41]采用微波促进, 以类似的n-Bu4NI/TsOH催化体系也能实现吲哚和亚磺酸作用合成吲哚硫醚39的反应.当吲哚的3-位被占据时, 2-号位的C—H键可发生反应得到吲哚-2-硫醚化合物.有趣的是, 同样采用芳基亚磺酸为硫源, Wang等[42]发现以水为反应介质时, 其和吲哚在室温下可以发生反应, 选择性地得到3-亚磺酰基吲哚53而不是上述催化方法中的吲哚硫醚.作者通过质谱监测, 发现了亚磺酰阳离子中间体54的存在.这一结果说明, 反应的可能的机理是亚磺酸在质子存在条件下通过脱水得到54, 54再和吲哚的亲核3-号位结合得到目标产物53 (Scheme 14).
 图式14
亚磺酸为硫源水相中吲哚3-C—H硫芳基化反应
图式14.
3-C—H Sulfenylation of indoles using sulfinic acids as thio sources in water
图式14
亚磺酸为硫源水相中吲哚3-C—H硫芳基化反应
图式14.
3-C—H Sulfenylation of indoles using sulfinic acids as thio sources in water
在吲哚的C—H硫醚化反应中另一个有效的硫源是N-硫代邻苯二甲酰亚胺55. Tudge等[43]采用55和吲哚进行反应, 使用溴化镁为催化剂, 在N, N-二甲基乙酰胺为介质, 在90 ℃加热条件下顺利实现了吲哚硫醚化合物的合成, 该反应中主要起催化作用的为卤素离子, 因其可以和54作用原位产生RS-X活性中间体, 进而发生硫醚化反应(Eq. 6).
Cossy等[44]采用结构类似的酰亚胺硫源5和吲哚反应, 采用过量的三氟乙酸为促进剂, 在室温条件下以及二氯甲烷溶剂中实现了吲哚2-号位的C—H硫醚化反应得到产物吲哚-2-硫醚51.反应具有较好的适用范围, 对多种3-位未被占据的吲哚都能高效转化.根据作者推测, 这一有别于大多数吲哚硫醚化反应的选择性结果主要是因为三氟乙酸的存在.反应第一步生成吲哚-3-硫醚, 而三氟乙酸引发的后续加成、重排和消除等串联转化最终得到目标产物51 (Eq. 7).
在无过渡金属催化的吲哚的C—H硫醚化反应中, 被用作硫源的还有Bunte盐(硫代硫酸酯钠盐). Luo等[45]以Bunte盐55为硫源和吲哚反应, 在催化量的碘存在下以DMSO同时作为氧化剂和溶剂, 顺利实现了吲哚-3-硫醚类化合物的合成(Eq. 8).同年, Wang和Ji等[46]采用四丁基碘化胺为促进剂, 在二氯乙烷(DCE)中加热也实现同样的反应(Eq. 8).两种催化方法在底物适用范围上略有差异, 但主要反应机理都涉及到烃基硫碘活性中间体.
除了吲哚之外, 其它多种已知的富电子芳杂环化合物的C—H硫醚化反应研究在过去的几年时间里也取得了较大进展.例如, Lu等[47]以碘为催化剂, 1, 4-二氧六环为溶剂, 实现了3-取代/2-取代苯并呋喃类化合物56/58和芳基磺酰肼之间的C—H键硫醚化反应, 建立了合成苯并呋喃-2-硫醚/苯并呋喃-3-硫醚57/59的方法(Scheme 15).
 图式15
苯并呋喃的2-和3-C—H硫芳基化反应
图式15.
2-and 3-C—H sulfenylation of benzofurans
图式15
苯并呋喃的2-和3-C—H硫芳基化反应
图式15.
2-and 3-C—H sulfenylation of benzofurans
Zhao等[48]近期以KI/PPh3为催化体系建立了类似的2-芳基/3-芳基苯并呋喃56/58的C-2和C-3硫醚化反应, 该方法中芳基磺酰氯被用做硫化试剂, 在EtOH中加热进行.同时, 该催化方法可用于吡唑啉酮类化合物60的C—H硫醚化反应合成硫醚产物61 (B, Scheme 16)[49]. Wei和Wang等[50]通过研究发现, 采用硫酚作为硫源和60在NaOH促进下也能和60发生C—H键硫醚化反应得到产物61.
 图式16
苯并呋喃的2-和3-以及吡唑酮的C—H硫芳基化反应
图式16.
2-/3-C—H sulfenylation of benzofurans and C—H sulfenylation of pyrazolones
图式16
苯并呋喃的2-和3-以及吡唑酮的C—H硫芳基化反应
图式16.
2-/3-C—H sulfenylation of benzofurans and C—H sulfenylation of pyrazolones
Bolm课题组[51]在2012年以对甲苯基二硫醚为硫源, Cs2CO3为碱, 1, 4-二氧六环为溶剂, 在反应温度130 ℃下, 完成了苯并杂环62(苯并噻唑, 苯并咪唑, 吲哚等)以及1, 3, 4-噁二唑65的C—H键硫醚化反应, 高产率的合成了多种芳杂环硫醚类化合物63、64和66 (Scheme 17). Braga等[52]采用K2CO3为碱性促进剂, 在DMSO中100 ℃加热也能实现芳杂环65的C—H硫醚化反应得到产物66, 且该催化方法对二硒醚类底物反应也具有优秀的适用性.
 图式17
二硫醚为硫源各种芳杂环的C—H硫杂环化合物的C—H硫芳基化反应
图式17.
C—H sulfenylation of various heteroaryls using disulfides as thio sources
图式17
二硫醚为硫源各种芳杂环的C—H硫杂环化合物的C—H硫芳基化反应
图式17.
C—H sulfenylation of various heteroaryls using disulfides as thio sources
You课题组[53]以三苯基膦为催化剂, 在甲苯中120 ℃加热实现了另一种芳杂环化合物吲嗪67的C—H硫醚化反应.该合成方法中苯磺酰氯作为硫源, 无需金属催化剂可合成得到吲嗪-3-硫醚类化合物68.同时, 作为芳香C—H键供体, 吲哚和富电子苯衍生物在该反应体系中也展示了良好兼容性.机理方面, 作者推测PPh3作为还原剂先将磺酰氯直接还原为芳基硫氯这一活性中间体, 该中间体作为硫亲电试剂和吲嗪通过亲电取代反应得到目标产物.反应的选择性由芳杂环中的氮原子定位进行控制(Scheme 18).
 图式18
吲嗪的3-C—H硫芳基化反应
图式18.
3-C—H sulfenylation of indolizines
图式18
吲嗪的3-C—H硫芳基化反应
图式18.
3-C—H sulfenylation of indolizines
Adimurthy等[54]以N-氯代丁二酰亚胺(NCS)为促进剂, 1, 2-二氯甲烷(DCM)为溶剂, 在室温下实现了咪唑[1, 2-a]并吡啶类化合物69的C—H硫醚化反应, 反应在咪唑环上的C—H键进行, 得到产物70.同样反应条件下, 具有类似结构的咪唑并环化合物71也可以进行相应的C—H硫醚化反应, 得到产物为72 (Scheme 19). Hiebel等[55]采用另一不同的无金属催化体系, 即I2为催化剂, H2O2为氧化剂的催化体系, 在PEG40介质中及温和的加热条件下也可完成类似的以硫酚为硫源, 咪唑[1, 2-a]并吡啶的C—H硫醚化反应.同时, 该催化体系可以扩展到类似门咪唑并硫源氮杂环化合物73, 通过硫醚化反应得到相应的目标产物硫醚74.在这一反应中, H2O2将硫酚直接氧化成二硫醚, 二硫醚和碘反应后得到硫碘活性亲电中间体, 该中间体和咪唑[1, 2-a]并吡啶通过加成-消除的芳基碳-氢键亲电取代反应过程得到杂环硫醚目标产物, 同时释放出的HI在双氧水氧化作用下转化为分子碘并重复催化循环过程完成反应(Scheme 20).
 图式19
NCS促进的咪唑[1, 2-a]并吡啶C—H硫醚化反应
图式19.
NCS-promoted C—H thioetherification of imidazo[1, 2-a]pyridines
图式19
NCS促进的咪唑[1, 2-a]并吡啶C—H硫醚化反应
图式19.
NCS-promoted C—H thioetherification of imidazo[1, 2-a]pyridines
 图式20
碘催化下咪唑[1, 2-a]并吡啶及类似N-杂芳环的C—H硫醚化反应
图式20.
Iodine-catalyzed C—H thioetherification of imidazo[1, 2-a]pyridines and their aza-analogs
图式20
碘催化下咪唑[1, 2-a]并吡啶及类似N-杂芳环的C—H硫醚化反应
图式20.
Iodine-catalyzed C—H thioetherification of imidazo[1, 2-a]pyridines and their aza-analogs
Zhang等[56]则采用了二硫醚作为硫源, 离子液为介质, 碳酸铯作为促进剂, 在80 ℃加热条件下实现了咪唑[1, 2-a]并吡啶69的C—H硫醚化反应, 得到产物70.对于吲哚和吡咯, 该硫醚化反应也能顺利进行, 得到相应的芳杂环硫醚39和75 (Scheme 21).随后, Hajra等[57]以磺酰肼为硫源和吡啶[1, 2-a]并咪唑及类似杂环75进行反应, 在碘为催化剂, 乙醇为溶剂, 70 ℃加热条件反应10 h即可顺利进行芳基C—H键硫醚化反应, 得到类似的硫醚类化合物76.反应产率良好, 且芳基/烷基磺酰基都可以发生, 咪唑并[2, 1-b]噻唑也作为富电子芳香底物也具有很好的适用性(Scheme 22). Prabhu等[58]采用I2/DMSO催化体系, 苯硫酚及芳杂环硫酚77为硫源, 合成得到一系列双芳杂环硫醚78 (Scheme 22). Ge和Li等[59]以亚磺酸钠为硫化试剂, 在I2/PPh3催化体系下也建立了基于芳杂环底物C—H键硫醚化反应合成化合物78的方法(Scheme 22).最近, Braga等[60]采用二硫醚以及二硒醚79分别作为硫化和硒化试剂, 同样以为I2/DMSO催化体系, 系统研究并实现了咪唑[1, 2-a]并吡啶的芳基C—H键硫化和硒化合成产物80的反应.同样的C—H键转化在该体系中证实也适用于吲唑、苯并噻唑、苯并噁唑、苯并咪唑以及1, 3, 4-噁二唑等芳杂环底物, 合成得到相应硫醚/硒醚81 (Scheme 22). Lin等[61]最近报道了I2/PPh3催化体系促进下, 芳基磺酰氯作为硫化试剂同样可以有效实现包括吡啶[1, 2-a]并咪唑、吡唑酮和富电子的苯酚衍生物之间的C—H硫醚化反应合成多种硫醚类产物.
 图式21
二硫醚为硫源的咪唑[1, 2-a]并吡啶等杂环的C—H硫醚化反应
图式21.
Disulfides as thio sources for the C—H thioetherification of imidazo[1, 2-a]pyridines etc.
图式21
二硫醚为硫源的咪唑[1, 2-a]并吡啶等杂环的C—H硫醚化反应
图式21.
Disulfides as thio sources for the C—H thioetherification of imidazo[1, 2-a]pyridines etc.
 图式22
咪唑[1, 2-a]并吡啶和不同硫源的C—H硫醚化反应
图式22.
C—H thioetherification of imidazo[1, 2-a]pyridines with different thio sources
图式22
咪唑[1, 2-a]并吡啶和不同硫源的C—H硫醚化反应
图式22.
C—H thioetherification of imidazo[1, 2-a]pyridines with different thio sources
不仅咪唑并芳环类化合物的C—H键可以直接进行各种无金属催化下的硫醚化反应, 含富电子侧链取代的咪唑衍生物也可以发生类似的C—H官能化转化. Wang研究小组[62]以二硫醚为硫化试剂, 实现了碘促进下的环状饱和硫侧链取代的咪唑并类化合物82的C-4硫醚化反应, 得到了系列咪唑硫醚类化合物83.反应条件简单, 且对烷基及芳基二硫醚底物都适用(Eq. 9).
Yang等[63]报道了以碘为催化剂条件下常规吡唑类化合物84和硫酚/硫醇的脱氢C—H偶联硫醚化反应, 高效地合成了一系列4-硫醚类吡唑衍生物85, 反应在100 ℃加热条件下进行, 所使用的DMSO起到溶剂及氧化剂的双重作用, 不需要加入其它碱、配体等添加剂.该方法的重要之处在于将之前主要以吡唑酮为底物合成羟基吡唑硫醚的反应扩展到普通的吡唑衍生物的反应(Eq. 10).
作为另外一种重要的含硫官能团, 硫氰基的存在对于许多有机化合物的性质具有重要作用, 因此在化合物结构中引入硫氰基的合成方法研究也广受关注. 2015年, Hajra课题组先后报道了吡啶[1, 2-a]并咪唑及类似杂环69/富和Wang电子芳杂环71的硫氰基化反应.如Scheme 23所示, Hajra等[64]以NH4SCN为硫氰基源, 在室温, 蓝色LED灯光照, eosin Y为光催化剂和空气氧化下实现反应. Wang等[65]则以KSCN为硫氰基源, 在室温以及过硫酸钾为氧化剂的条件下实现, 同时该催化反应对富电子的苯衍生物如N, N-二甲基氨基苯也使用, 转化反应发生在氨基对位的C—H键上.
 图式23
咪唑[1, 2-a]并吡啶的C—H硫氰基化反应
图式23.
C—H thiocyanation of imidazo[1, 2-a]pyridines
图式23
咪唑[1, 2-a]并吡啶的C—H硫氰基化反应
图式23.
C—H thiocyanation of imidazo[1, 2-a]pyridines
Yi等[66]发展了以氟烷基磺酰氯91为氟烷基硫化试剂对吲哚38和吡咯90等芳杂环化合物进行氟烷基硫醚化反应的方法.该反应中亚磷酸二乙酯被用作还原剂, 和氟烷基磺酰氯原位发生氧化还原反应产生活性的氟烷基硫氯94, 继而在无金属催化条件下, 乙腈为介质, 加热至90 ℃顺利实现了氟烷基硫醚产物92和93的合成(Scheme 24).
 图式24
吲哚和吡咯的C—H多氟烷基硫醚化反应
图式24.
Perfluoroalkyl thiolation on the C—H bond of indoles and pyrroles
图式24
吲哚和吡咯的C—H多氟烷基硫醚化反应
图式24.
Perfluoroalkyl thiolation on the C—H bond of indoles and pyrroles
在直接C—H硫醚化反应的基础上, 通过一步三组分反应直接同时完成芳杂环的构建以及串联C—H转化反应也成为一种实用的合成双芳基硫醚的方法. Guo等[67]报道了多取代吡唑硫醚的串联合成方法.该方法采用α-氰基酮95或乙酰丙酮96和芳基肼97以及芳基磺酰肼在碘催化下反应, 其中亚甲基酮类化合物和芳基肼在反应中作为底物构建吡唑环, 而磺酰肼作为硫源在碘催化下和原位形成的吡唑发生C—H硫醚化反应, 得到产物98和99, 两类产物的生成条件稍有不同(Scheme 25).同年, 该课题组[68]还报道了从底物95和磺酰肼出发合成吡唑并吡啶类化合物C—H键硫醚化的方法.该合成也在碘催化下进行, 首先是两分子的95在碘催化下和磺酰肼反应, 经过NaOH处理得到吡唑并吡啶类化合物100, 该中间体结构中的两个芳基C—H键均可以在碘催化下和磺酰肼发生硫醚化反应, 最终得到目标产物101 (Scheme 26).
 图式25
三组分C—H硫芳基化反应合成吡唑硫醚
图式25.
Three-component C—H sulfenylation reactions for the synthesis of pyrazole thioethers
图式25
三组分C—H硫芳基化反应合成吡唑硫醚
图式25.
Three-component C—H sulfenylation reactions for the synthesis of pyrazole thioethers
 图式26
串联C—H硫醚化反应合成吡唑并吡啶硫醚
图式26.
Tandem C—H thioetherification for the synthesis of pyrazolo[1, 5-a]pyridines
图式26
串联C—H硫醚化反应合成吡唑并吡啶硫醚
图式26.
Tandem C—H thioetherification for the synthesis of pyrazolo[1, 5-a]pyridines
Basu等[69]采用2-氨基吡啶102, 苯乙酮103和硫酚/硫醇进行三祖分反应, 在氧化石墨烯催化剂和NaI存在条件下, 通过在甲苯中和空气氛围下加热至80 ℃直接得到了硫醚类产物70.这一合成过程中也是首先发生2-氨基吡啶和苯乙酮之间构建吡啶[1, 2-a]并咪唑杂环中间体的反应, 然后通过C—H硫醚化得到目标产物(Eq. 11).
2 非芳基C(sp2)—H键硫醚化反应
作为另外一种典型的C(sp2)—H键, 烯基上的C—H键和芳环上的C—H键在偶联化学中具有一些共性.尽管目前已知的绝大多数烯基C—H键偶联官能化反应都通过过渡金属催化实现, 无金属催化的相关C—H偶联反应在近几年也已经有一些进展. Wang等[70]在2014年报道了烯基官能化二硫缩烯酮104在碘催化下通过分子内的烯基C—H键硫醚化反应得到噻吩酮106的方法, 反应中得到少量的六元杂环副产物107.在相同条件下, 二甲硫基苯基烯酮105也可发生类似的C—H硫醚化反应得到产物108, 同时产生副产物109 (Scheme 27).
 图式27
二硫缩烯酮分子类C—H硫化反应
图式27.
Intramolecular C—H thiolation of thioacetal ketenes
图式27
二硫缩烯酮分子类C—H硫化反应
图式27.
Intramolecular C—H thiolation of thioacetal ketenes
最近, Wan等[71]成功发展了烯胺酮类化合物的α-C—H键和硫酚的芳基硫醚化反应, 该方法以简单的钾盐KIO3为催化剂, 生物质绿色溶剂乳酸乙酯为介质, 在空气氛围以及90 ℃加热条件下即可实现.值得一提的是, 不论是对于NH2, NH还是N, N-双取代氨基结构以及具有环状或链状结构的烯胺酮类化合物110/111, 该催化体系都展示了良好的兼容性, 顺利得到目标产物112/ 113, 是一条较为通用的简单合成烯胺硫醚的新途径.根据相关控制反应的结果, 作者推测反应首先是通过碘酸跟踪的高价碘对烯胺酮双键中的α-碳-氢键进行氧化得到含五价碘的烯胺酮中间体.该中间体和原位氧化形成的二硫醚类化合物发生基于硫-硫键断裂的置换转化得到烯胺酮硫醚产物, 同时产生二氧化碘硫中间体.二氧化碘硫中间体和反应原位产生的氢氧化钾作用可以再次转化为碘酸根离子, 促进反应的循环进行(Scheme 28).
 图式28
KIO3催化的烯胺酮C—H硫芳基化反应
图式28.
KIO3-catalyzed C—H sulfenylation of enaminones
图式28
KIO3催化的烯胺酮C—H硫芳基化反应
图式28.
KIO3-catalyzed C—H sulfenylation of enaminones
以该催化C—H键硫醚化反应为基础, 该小组[72]最近采用邻羟基苯基烯胺酮113为起始底物和硫酚类化合物反应, 在类似的催化体系以及更加温和的加热条件下建立了串联C—H键硫醚化以及C—N键断裂构建C—O关环转化直接合成3-芳硫基色酮类化合物114的新方法.控制反应结果显示反应过程可能是113和硫酚先发生C—H硫醚化反应得到中间体115, 该中间体再原位发生C—O键关环反应得到目标产物(Scheme 29).
 图式29
串联烯胺酮C—H硫芳基化反应合成3-色酮硫醚
图式29.
Tandem enaminone C—H sulfenylation for the synthesis of chromone-3-thioethers
图式29
串联烯胺酮C—H硫芳基化反应合成3-色酮硫醚
图式29.
Tandem enaminone C—H sulfenylation for the synthesis of chromone-3-thioethers
Zhou研究小组[73]以NH4I为促进剂, 磺酰肼为硫源, 在二甲基乙酰胺溶剂中实现了色酮116的3-位C—H键的硫醚化反应, 反应在135 ℃加热条件下进行, 所得产物也为色酮硫醚114; 同时, 将反应溶剂更换为乙腈, 以DMSO为替代硫源, 可以得到甲基硫醚化的色酮117 (Scheme 30).随后, 该小组更换磺酰氯为硫源, DMF为反应为介质, 同样在NH4I存在以及135 ℃加热条件下实现了116的C—H硫醚化生成114的反应, 该反应对烷基以及芳基磺酰氯都适用[74].此外, 该课题组还报道了采用硫酚为硫化试剂, 在NH4I/DMF体系以及135 ℃加热下和116反应得到产物114的方法[75].
 图式30
色酮3-C—H硫醚化反应
图式30.
3-C—H thioetherification of chromones
图式30
色酮3-C—H硫醚化反应
图式30.
3-C—H thioetherification of chromones
最近, Chen和Zhou等[76]报道了一例无金属催化条件下α, β-不饱和醛类化合物118中烯基C—H键的硫醚化反应.在卡宾前体120的催化下, 通过碱的作用原位产生活泼的卡宾催化剂, 在室温即可实现118中烯键上C—H键的硫醚化反应得到产物119.该反应中所用硫醚化试剂为N-硫代邻苯二甲酰亚胺.反应过程首先是亲核卡宾催化剂或N-硫代酰亚胺中的亲核片段对碳-碳双键进行加成反应得到碳负离子中间体, 该碳负离子和N-硫代酰亚胺中的亲电性硫位点结合, 并继续发生消除亲核试剂的反应得到目标产物(Scheme 31).
 图式31
N-杂卡宾催化的烯醛α-C—H硫芳基化反应
图式31.
NHC-catalyzed α-C—H sulfenylation of enals
图式31
N-杂卡宾催化的烯醛α-C—H硫芳基化反应
图式31.
NHC-catalyzed α-C—H sulfenylation of enals
3 总结和展望
从前文研究进展可以看出, 作为构建C(sp2)—S键的一种新型方法, 无过渡金属催化的C(sp2)—H硫醚化反应已经引起了广泛的关注, 这类方法本身不需过渡金属催化剂以及直接在C—H键进行转化的特性, 在可持续性和环境友好方面具有不可比拟的优势.同时, 过去的10来多年时间里, 这类合成方法的研究无疑也取得了长足的发展, 特别是在新型硫源的开发以及不同催化剂模式的发展方面均已有丰富的文献报道.同时, 需要看到的是, 该研究领域当前仍然面临着一些挑战, 其中主要的问题就是C—H给体的通用性问题.从已知文献可以发现, 不论是非杂环芳香化合物、杂环芳香化合物还是烯基C—H键底物, 富电子或者高度极化的结构是绝大多数底物的共性, 对于非极性或极性较低的C—H键给体, 如不含强给电子基或缺点子的芳环以及烯烃, 目前要实现相应的无过渡金属硫醚化反应还比较困难, 这无疑也将成为未来本领域研究的重要课题.
-
-
[1]
(a) Felix, A. M.; Unowsky, J.; Bontempo, J.; Fryer, R. I. J. Med. Chem. 1968, 11, 929.
(b) Gangjee, A.; Zeng, Y.; Talreja, T.; McGuire, J. J.; Kisliuk, R. L.; Queener, S. F. J. Med. Chem. 2007, 50, 3046.
(c) Pedras, M. S. C.; Zheng, Q.-A.; Strelkov, S. J. Agric. Food Chem. 2008, 56, 9949.
(d) Halim, M.; Yee, D. J.; Sames, D. J. Am. Chem. Soc. 2008, 130, 14123.
(e) Wilson, A. J.; Kerns, J. K.; Callahan, J. F.; Moody, C. J. J. Med. Chem. 2013, 56, 7463. -
[2]
(a) De Martino, G.; Edler, M. C.; La Regina, G.; Coluccia, A.; Barbera, M. C.; Barrow, D.; Nicholson, R. I.; Chiosis, G.; Brancale, A.; Hamel, E.; Artico, M.; Silvestri, R. J. Med.Chem. 2006, 49, 947.
(b) Ragno, R.; Coluccia, A.; La Regina, G.; De Martino, G.; Piscitelli, F.; Lavecchia, A.; Novellino, E.; Bergamini, A.; Ciaprini, C.; Sinistro, A.; Maga, G.; Crespan, E.; Artico, M.; Silvestri, R. J. Med. Chem. 2006, 49, 3172.
(c) Armer, R. E.; Wynne, G. M. WO 2008012511, 2008 [Chem. Abstr. 2008, 148, 183423]. -
[3]
For selected examples, see (a) Mann, G.; Baranano, D.; Hartwig, J. F.; Rheingold, A. L.; Guzei, I. A. J. Am. Chem. Soc. 1998, 120, 9205.
(b) Li, G. Angew. Chem., Int. Ed. 2001, 40, 1513.
(c) Wong, Y.-C.; Jayanth, T. T.; Cheng, C.-H. Org. Lett. 2006, 8, 5613.
(d) Rout, L.; Sen, T. K.; Punniyamurthy, T. Angew. Chem., Int. Ed. 2007, 46, 5583.
(e) Arisawa, M.; Suzuki, T.; Ishikawa, T.; Yamaguchi, M. J. Am. Chem. Soc. 2008, 130, 12214.
(f) Ma, D.; Cai, Q. Acc. Chem. Res. 2008, 41, 1450.
(g) Jiang, Z.; She, J.; Lin, X. Adv. Synth. Catal. 2009, 351, 2558.
(h) Wang, X.; Yang, F.; Xue, Z. Chin. J. Org. Chem. 2015, 35, 29.
(i) Zhao, F.; Jia, X.; Wang, D.; Fei, C.; Wu, C.; Wang, J.; Liu, H. Chin. J. Org. Chem. 2017, 37, 284. -
[4]
(a) Qian, P. ; Zhang, M. ; Hu, M. ; Cheng, J. J. Org. Chem. 2010, 75, 6732.
(b) Wendlandt, A. E. ; Suess, A. M. ; Stahl, S. S. Angew. Chem. , Int. Ed. 2011, 50, 11062.
(c) Tran, L. D. ; Popov, I. ; Daugulis, O. J. Am. Chem. Soc. 2012, 134, 18237.
(d) Dai, C. ; Xu, Z. ; Huang, F. ; Yu, Z. ; Gao, Y. -F. J. Org. Chem. 2012, 77, 4414.
(e) Rosario, A. R. ; Casola, K. K. ; Oliveira, C. E. S. ; Zeni, G. Adv. Synth. Catal. 2013, 355, 2960.
(f) Yin, W. ; Wang, Z. ; Huang, Y. Adv. Synth. Catal. 2014, 356, 2998.
(g) Saravanan, P. ; Anbarasan, P. Org. Lett. 2014, 16, 848.
(h) Lin, C. ; Li, D. ; Wang, B. ; Yao, J. ; Zhang, Y. Org. Lett. 2015, 17, 1328.
(i) Yang, K. ; Wang, Y. ; Chen, X. ; Kadi, A. A. ; H. -K. Fun, H. Sun, Y. Zhang, H. Lu, Chem. Commun. 2015, 51, 3582.
(j) Hu, Z. ; Tong, X. ; Liu, G. Chin. J. Org. Chem. 2015, 35, 539.
(k) Pei, P. ; Zhang, F. ; Yi, H. ; Lei, A. Acta Chim. Sinica 2017, 75, 15 (in Chinese).
(裴朋昆, 张凡, 易红, 雷爱文, 化学学报, 2017, 75, 15. ) -
[5]
Prasad, C. D.; Balkrishna, S. J.; Kumar, A.; Bhakuni, B. S.; Shrimali, S. B.; Kumar, S. J. Org. Chem. 2013, 78, 1434. doi: 10.1021/jo302480j
-
[6]
Tian, H.; Zhu, C.; Yang, H.; Fu, H. Chem. Commun. 2014, 50, 8875. doi: 10.1039/C4CC03600J
-
[7]
Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D. G.; Cossy, J. Org. Lett. 2015, 17, 3898. doi: 10.1021/acs.orglett.5b01889
-
[8]
Yang, D.; Yan, K.; Wei, W.; Zhao, J.; Zhang, M.; Sheng, X.; Li, G.; Lu, S.; Wang, H. J. Org. Chem. 2015, 80, 6083. doi: 10.1021/acs.joc.5b00540
-
[9]
Yan, K.; Yang, D.; Sun, P.; Wei, W.; Liu, Y.; Li, G.; Lu, S.; Wang, H. Tetrahedron Lett. 2015, 56, 4792. doi: 10.1016/j.tetlet.2015.06.057
-
[10]
Parumala, S. K. R.; Peddinti, R. K. Green Chem. 2015, 17, 4068. doi: 10.1039/C5GC00403A
-
[11]
Blanco, G. A.; Baumgartner, M. T. Tetrahedron Lett. 2011, 52, 7061. doi: 10.1016/j.tetlet.2011.10.053
-
[12]
Varun, B. V.; Prabhu, K. R. J. Org. Chem. 2014, 79, 9655. doi: 10.1021/jo501793q
-
[13]
Kang, X.; Yan, R.; Yu, G.; Pang, X.; Liu, X.; Li, X.; Xiang, L.; Huang, G. J. Org. Chem.2014, 79, 10605. doi: 10.1021/jo501778h
-
[14]
Zhao, X.; Li, T.; Zhang, L.; Lu, K. Org. Biomol. Chem. 2016, 14, 1131. doi: 10.1039/C5OB02193F
-
[15]
Wang, D.; Zhang, R.; Lin, S.; Yan, Z.; Guo, S. RSC Adv. 2015, 5, 108030. doi: 10.1039/C5RA24351C
-
[16]
Wang, D.; Guo, S.; Zhang, R.; Lin, S.; Yan, Z. RSC Adv. 2016, 6, 54377. doi: 10.1039/C6RA02302A
-
[17]
Zhao, X.; Deng, Z.; Wei, A.; Li, B.; Lu, K. Org. Biomol. Chem. 2016, 14, 7304. doi: 10.1039/C6OB01006G
-
[18]
Matsugi, M.; Gotanda, K.; Murata, K.; Kita, Y. Chem. Commun. 1997, 1387.
-
[19]
Matsugi, M.; Murata, K.; Gotanda, K.; Nambu, H.; Anikumar, G.; Matsumoto, K.; Kita, Y. J. Org. Chem. 2001, 66, 2434. doi: 10.1021/jo001710q
-
[20]
Schlosser, K. M.; Krasutsky, A. P.; Hamilton, H. W.; Reed, J. E.; Seaton, K. Org. Lett. 2004, 6, 819. doi: 10.1021/ol049956v
-
[21]
Yadav, J. S.; Reddy, B. V. S.; Reddy, Y. J. Tetrahedron Lett. 2007, 48, 7034. doi: 10.1016/j.tetlet.2007.07.130
-
[22]
Liu, Y.; Zhang, Y.; Hu, C.; Wan, J-P.; Wen, C. RSC Adv. 2014, 4, 35528. doi: 10.1039/C4RA05206D
-
[23]
He, Y.; Liu, S.; Wen. P.; Tian, W.; Ren, X.; Zhou, Q.; Ma, H.; Huang, G. ChemistrySelect 2016, 1, 1567. doi: 10.1002/slct.201600257
-
[24]
Saima; Equbal, D.; Lavekar, A. G.; Sinha, A. K. Org. Biomol. Chem. 2016, 14, 6111. doi: 10.1039/C6OB00930A
-
[25]
Zhang, X.; Zhou, X.; Xiao, H.; Li, X. RSC Adv. 2013, 3, 22280. doi: 10.1039/c3ra44484h
-
[26]
Zhou, X.; L, X. RSC Adv. 2014, 4, 1241.
-
[27]
Ge, W.; Wei, Y. Green Chem. 2012, 14, 2066. doi: 10.1039/c2gc35337g
-
[28]
Huang, D.; Chen, J.; Dan, W.; Ding, J.; Liu, M.; Wu, H. Adv. Synth. Catal. 2012, 354, 2123. doi: 10.1002/adsc.v354.11/12
-
[29]
Sang, P.; Chen, Z.; Zou, J.; Zhang, Y. Green Chem. 2013, 15, 2096. doi: 10.1039/c3gc40724a
-
[30]
Prasad, D. C.; Kumar, S.; Sattar, M.; Amit, A.; Kumar, S. Org. Biomol. Chem. 2013, 11, 8036. doi: 10.1039/c3ob41601a
-
[31]
Azeredo, J. B.; Godoi, M.; Martins, G. M.; Silveira, C. C.; Braga, A. L. J. Org. Chem. 2014, 79, 4125. doi: 10.1021/jo5000779
-
[32]
Xiao, F.; Xie, H.; Liu, S.; Deng, G.-J. Adv. Synth. Catal. 2014, 356, 364. doi: 10.1002/adsc.201300773
-
[33]
Katrun, P.; Hongthong, S.; Hlkhlai, S.; Pohmakotr, M.; Reutrakul, V.; Soorukram, D.; Jaipetch, T.; Kuhakarn, C. RSC Adv. 2014, 4, 18933. doi: 10.1039/c4ra02607a
-
[34]
Rao, H.; Wang, P.; Wang, J.; Li, Z.; Sun, X.; Cao, S. RSC Adv. 2014, 4, 49165. doi: 10.1039/C4RA08669D
-
[35]
Kumaraswamy, G.; Raju, R.; Narayanarao, V. RSC Adv. 2015, 5, 22718. doi: 10.1039/C5RA00646E
-
[36]
Zhao, X.; Lu, X.; Wei, A.; Jia. X.; Chen, J.; Lu, K. Tetrahedron Lett. 2016, 57, 5330. doi: 10.1016/j.tetlet.2016.10.053
-
[37]
Yang, F-L.; Tian, S-K. Angew. Chem., Int. Ed. 2013, 52, 4929. doi: 10.1002/anie.v52.18
-
[38]
Yang, Y.; Zhang, S.; Tang, L.; Hu, Y.; Zha, Z.; Wang, Z. Green Chem. 2016, 18, 2609. doi: 10.1039/C6GC00313C
-
[39]
Rahaman, R.; Devi, N.; Sarma, K.; Barman, P. RSC Adv. 2016, 6, 10873. doi: 10.1039/C5RA24851E
-
[40]
Liu, C-R.; Ding, L-H. Org. Biomol. Chem. 2015, 13, 2251. doi: 10.1039/C4OB02575J
-
[41]
Rahaman, R.; Devi, N.; Bhagawati, J. R.; Barman, P. RSC Adv. 2016, 6, 18929. doi: 10.1039/C5RA26425A
-
[42]
Miao, T.; Li, P.; Zhang, Y.; Wang, L. Org. Lett. 2015, 17, 832. doi: 10.1021/ol503659t
-
[43]
Tudge, M.; Tamiya, M.; Savarin, C.; Humphrey, G. R. Org. Lett. 2006, 8, 565. doi: 10.1021/ol052615c
-
[44]
Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D. G.; Cossy, J. Chem. Commun. 2015, 51, 13898. doi: 10.1039/C5CC05421D
-
[45]
Qi, H.; Zhang, T.; Wan, K.; Luo, M. J. Org. Chem. 2016, 81, 4262. doi: 10.1021/acs.joc.6b00636
-
[46]
Li, J.; Cai, Z.-J.; Wang, S.-Y.; Ji, S.-J. Org. Biomol. Chem. 2016, 14, 9384. doi: 10.1039/C6OB01528J
-
[47]
Zhao, X.; Zhang, L.; Lu, X.; Li, T.; Lu, K. J. Org. Chem. 2015, 80, 2918. doi: 10.1021/acs.joc.5b00146
-
[48]
Zhao, X.; Lu, X.; Wei, A.; Jia, X.; Chen, J.; Lu, K. Tetrahedron Lett. 2016, 57, 5330. doi: 10.1016/j.tetlet.2016.10.053
-
[49]
Zhao, X.; Zhang, L.; Li. T.; Liu, G.; Wang, H.; Lu, K. Chem. Commun. 2014, 50, 13121. doi: 10.1039/C4CC05237D
-
[50]
Liu, X.; Cui, H.; Yang, D.; Dai, S.; Zhang, T.; Sun, J.; Wei, W.; Wang, H. RSC Adv. 2016, 6, 51830. doi: 10.1039/C6RA09739A
-
[51]
Zhou, L-H.; Reball, J.; Mottweiler, J.; Bolm, C. Chem. Commun. 2012, 48, 11307. doi: 10.1039/c2cc36711d
-
[52]
Rafique, J.; Saba, S.; Rosário, A. R.; Zeni, G.; Braga, A. L. RSC Adv. 2014, 4, 51648. doi: 10.1039/C4RA10490K
-
[53]
Wu, Q.; Zhao, D.; Qin, X.; Lan, J.; You, J. Chem. Commun. 2011, 47, 9188. doi: 10.1039/c1cc13633j
-
[54]
Ravi, C.; Mohan, D. C.; Adimurthy, S. Org. Lett. 2014, 16, 2978. doi: 10.1021/ol501117z
-
[55]
Hiebel, M-A.; Berteina-Raboin, S. Green Chem. 2015, 17, 937. doi: 10.1039/C4GC01462F
-
[56]
Gao, Z.; Zhu, X.; Zhang, R. RSC Adv. 2014, 4, 19891. doi: 10.1039/C4RA01240B
-
[57]
Bagdi, A. K.; Mitra, S.; Ghosh, M.; Hajra, A. Org. Biomol. Chem. 2015, 13, 3314. doi: 10.1039/C5OB00033E
-
[58]
Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2016, 81, 7838. doi: 10.1021/acs.joc.6b01487
-
[59]
Huang, X.; Wang, S.; Li, B.; Wang, X.; Ge. Z.; Li, R. RSC Adv. 2015, 5, 22654. doi: 10.1039/C4RA17237J
-
[60]
Rafique, J.; Saba, S.; Rosário, A. R.; Braga, A. L. Chem. Eur. J. 2016, 22, 11854. doi: 10.1002/chem.201600800
-
[61]
Wang, D.; Zhang, R.; Lin, S.; Deng, R.; Yan, Z. Chin. J. Org. Chem. 2016, 36, 2757. doi: 10.6023/cjoc201604056
-
[62]
Liu, W.; Wang, S.; Chen, Y.; Huang, Y.; Li, Z.; Wang, A. Phosphorus Sulfur, Silicon Relat. Elem. 2016, 191, 689. doi: 10.1080/10426507.2015.1073278
-
[63]
Yang, D.; Sun, P.; Wei, W.; Meng, L.; He, L.; Fang, B.; Jiang, W. Org. Chem. Front. 2016, 3, 1457. doi: 10.1039/C6QO00407E
-
[64]
Mitra, S.; Ghosh, M.; Mishra, S.; Hajra, A. J. Org. Chem. 2015, 80, 8275. doi: 10.1021/acs.joc.5b01369
-
[65]
Yang, D.; Yan, K.; Wei, W.; Li, G.; Lu, S.; Zhao, C.; Tian, L.; Wang, H. J. Org. Chem. 2015, 80, 11073. doi: 10.1021/acs.joc.5b01637
-
[66]
Jiang, L.; Yi, W.; Liu, Q. Adv. Synth. Catal. 2016, 358, 3700. doi: 10.1002/adsc.201600651
-
[67]
Sun, J.; Qiu, J.-K.; Zhu, Y.-L.; Guo, C.; Hao, W.-J.; Jiang, B.; Tu, S.-J. J. Org. Chem. 2015, 80, 8217. doi: 10.1021/acs.joc.5b01280
-
[68]
Sun, J.; Qiu, J.-K.; Jiang, B.; Hao, W.-J.; Guo, C.; Tu, S.-J. J. Org. Chem. 2015, 80, 3321. doi: 10.1021/jo502912m
-
[69]
Kundu, S.; Basu, B. RSC Adv. 2015, 5, 50178. doi: 10.1039/C5RA04983K
-
[70]
Zheng, G.; Ma, X.; Liu, B.; Dong, Y.; Wang, M. Adv. Synth. Catal. 2014, 356, 743. doi: 10.1002/adsc.v356.4
-
[71]
Wan, J.-P.; Zhong, S.; Xie, L.; Cao, X.; Liu, Y.; Wei, L. Org. Lett. 2016, 18, 584. doi: 10.1021/acs.orglett.5b03608
-
[72]
Zhong, S.; Liu, Y.; Cao, X; Wan, J.-P. ChemCatChem 2017, 9, 465. doi: 10.1002/cctc.v9.3
-
[73]
Zhao, W.; Xie, P.; Bian, Z.; Zhou, A.; Ge, H.; Zhang, M.; Ding, Y.; Zheng, L. J. Org. Chem. 2015, 80, 9167. doi: 10.1021/acs.joc.5b01602
-
[74]
Zhao, W.; Zhou, A. ChemCatChem 2015, 7, 3464. doi: 10.1002/cctc.v7.21
-
[75]
Zhao, W.; Xie, P.; Bian, Z.; Zhou, A.; Ge, H.; Niu, B.; Ding, Y. RSC Adv. 2015, 5, 59861. doi: 10.1039/C5RA10763F
-
[76]
Dong, Y.-T.; Jin, Q.; Zhou, L.; Chen, J. Org. Lett. 2016, 18, 5708. doi: 10.1021/acs.orglett.6b02939
-
[1]
-

图式1 K2S2O8催化富电子苯的C—H硫/硒芳基化反应
Scheme 1 K2S2O8-catalyzed C—H sulfenylation/selenation of electron rich benzenes using disulfides/diselenides

图式2 N-硫代丁二酰亚胺为硫源的富电子苯C—H硫芳基化反应
Scheme 2 N-Thiobutanimides as thio sources for the sulfenylation of electron rich benzenes

图式3 硫酚/硫醇为硫源的富电子苯C—H硫芳基化反应
Scheme 3 Thiophenols/thiols as thio sources for the sulfenylation of electron rich benzenes

图式4 碘催化富电子苯和极性烯基C—H硫芳基化反应
Scheme 4 I2-catalyzed C—H sulfenylation of electron rich benzenes polor vinyls

图式5 K2S2O8/TfOH促进的苯基醚和吲哚C—H硫芳基化反应
Scheme 5 K2S2O8/TfOH-promoted C—H sulfenylation of phenyl ethers and indoles

图式6 磺酰肼为硫源的富电子苯/萘的C—H硫醚化反应
Scheme 6 Sulfonyl hydrazine as thiol sources in the C—H thiolation of electron benzenes/naphthalenes

图式7 亚磺酸钠和磺酰氯为硫源的富电子苯/萘的C—H硫醚化反应
Scheme 7 Sodium sulfinates and sufonyl chlorides as thiol sources in the C—H thiolation of electron benzenes/naphthalenes

图式8 O, S-缩醌酮为硫源的吲哚和富电子烯醚/苯基醚的C—H硫醚化反应
Scheme 8 Quinine O, S-acetals as thiol sources for the C—H thioetherification of indoles, electron rich vinyl/phenyl ethers

图式9 二硫醚为硫源的吲哚3-C—H硫醚化反应
Scheme 9 Disulfides as thio sources for the 3-C—H thioetherification of indoles

图式10 K2CO3催化下二硫醚和吲哚的3-C—H硫芳基化反应
Scheme 10 K2CO3-catalyzed indole 3-C—H sulfenylation using disulfides

图式11 过硫酸铵促进二硫醚为硫源的吲哚3-以及2, 3-二硫芳基化反应
Scheme 11 (NH4)2S2O8-promoted 3-and 2, 3-(di)sulfenylation of indoles using disulfides

图式12 亚磺酸钠为硫源的吲哚3-C—H硫醚化反应
Scheme 12 3-C—H thioetherification of indoles using sodium sulfinates as thio sources

图式13 磺酰肼为硫源的吲哚3-和2-C—H硫醚化反应
Scheme 13 3-and 2-C—H Thioetherification of indoles using sulfonyl hydrazines as thio sources

图式14 亚磺酸为硫源水相中吲哚3-C—H硫芳基化反应
Scheme 14 3-C—H Sulfenylation of indoles using sulfinic acids as thio sources in water

图式16 苯并呋喃的2-和3-以及吡唑酮的C—H硫芳基化反应
Scheme 16 2-/3-C—H sulfenylation of benzofurans and C—H sulfenylation of pyrazolones

图式17 二硫醚为硫源各种芳杂环的C—H硫杂环化合物的C—H硫芳基化反应
Scheme 17 C—H sulfenylation of various heteroaryls using disulfides as thio sources

图式19 NCS促进的咪唑[1, 2-a]并吡啶C—H硫醚化反应
Scheme 19 NCS-promoted C—H thioetherification of imidazo[1, 2-a]pyridines

图式20 碘催化下咪唑[1, 2-a]并吡啶及类似N-杂芳环的C—H硫醚化反应
Scheme 20 Iodine-catalyzed C—H thioetherification of imidazo[1, 2-a]pyridines and their aza-analogs

图式21 二硫醚为硫源的咪唑[1, 2-a]并吡啶等杂环的C—H硫醚化反应
Scheme 21 Disulfides as thio sources for the C—H thioetherification of imidazo[1, 2-a]pyridines etc.

图式22 咪唑[1, 2-a]并吡啶和不同硫源的C—H硫醚化反应
Scheme 22 C—H thioetherification of imidazo[1, 2-a]pyridines with different thio sources

图式24 吲哚和吡咯的C—H多氟烷基硫醚化反应
Scheme 24 Perfluoroalkyl thiolation on the C—H bond of indoles and pyrroles

图式25 三组分C—H硫芳基化反应合成吡唑硫醚
Scheme 25 Three-component C—H sulfenylation reactions for the synthesis of pyrazole thioethers

图式26 串联C—H硫醚化反应合成吡唑并吡啶硫醚
Scheme 26 Tandem C—H thioetherification for the synthesis of pyrazolo[1, 5-a]pyridines

图式29 串联烯胺酮C—H硫芳基化反应合成3-色酮硫醚
Scheme 29 Tandem enaminone C—H sulfenylation for the synthesis of chromone-3-thioethers
-


 下载:
下载:






























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 17
- 文章访问数: 3320
- HTML全文浏览量: 475
 下载:
下载:
