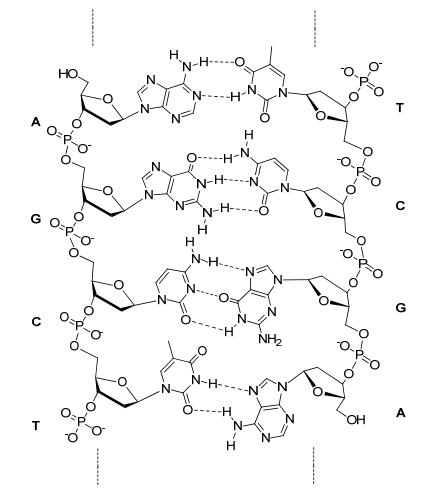
 图式 1
DNA分子的双股结构
Scheme1.
Double strand structure of DNA
图式 1
DNA分子的双股结构
Scheme1.
Double strand structure of DNA
引用本文:
朱磊, 李博解, 严沣, 汪连生. 人工合成类DNA双股高分子的研究进展[J]. 有机化学,
2017, 37(11): 2800-2817.
doi:
10.6023/cjoc201604052

Citation: Zhu Lei, Li Bojie, Yan Feng, Wang Liansheng. Progress in the Synthesis of DNA-Like Double Strand Polymers[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2800-2817. doi: 10.6023/cjoc201604052
Citation: Zhu Lei, Li Bojie, Yan Feng, Wang Liansheng. Progress in the Synthesis of DNA-Like Double Strand Polymers[J]. Chinese Journal of Organic Chemistry, 2017, 37(11): 2800-2817. doi: 10.6023/cjoc201604052
人工合成类DNA双股高分子的研究进展
English
Progress in the Synthesis of DNA-Like Double Strand Polymers
Abstract:
Deoxyribonucleic acid (DNA) is the genetic material determining the makeup of all living cells and many viruses. DNA has a self-replicating feature which consists of two polynucleotide chains in the form of a double helix. The synthesis of DNA-like double strand polymers has attracted much attention over recent years due to the development of self-assembly chemistry and organic synthesis technology. The synthesis of double strand polymers divided by the interaction including non-covalent bonding and covalent bonding between linker and backbone is sumarized. Poly-norbornene which could be performed as effcient backbone is also described. A series of isotactic double strand polymeric ladderphanes have been successfully acheived based on such poly-norbornene backbone.
-
DNA分子即脱氧核糖核酸, 是染色体的重要组成成分, 同时也是组成基因的材料. 1953年, Watson和Crick[1]首次提出DNA分子的双股螺旋结构, 脱氧核糖与磷酸分子以酯键相连组成主链, 排列于外侧, 而内侧则由碱基对以氢键相连(Scheme 1).基于其双股的结构, DNA分子具有许多独特的性质, 如氧化还原性和导磁性, 在骨架上的磷酸基带有负电荷, 电子可在不同的碱基层之间发生跃迁[2~4], 使得DNA分子可以做为半导体[2b, 3]或超导体[4].而对于DNA分子最重要的研究, 是其精确的复制过程[5], 在聚合酶催化下, 一条单股的DNA分子可以精确控制另一股DNA分子的序列, 从而完成遗传信息的传递.早期相关的研究由Orgel[6]提出, 之后von Kiedrowski[7]利用核酸类似的分子进行研究, 而有机化学家则着重利用自组装[8]、模板效应[7, 9~12]及非共价键合成[9]等方法对复制过程进行模拟和研究.然而, 经过多年的努力, 除天然的核酸和氨基酸系统外, 对复制过程的研究仍停留在小分子阶段无法突破, 因此人工合成出具有DNA类似双股结构的高分子成为亟待解决的问题.
图式 1
DNA分子的双股结构
Scheme1.
Double strand structure of DNA
目前, 传统聚合方法所得到的高分子与天然生物高分子, 如蛋白质、核酸等, 存在着很大的差异(表 1).例如, 生物高分子的分子量分布PDI (polydispersity index)为1, 而合成高分子则大于1;生物高分子具有规则排列的非重复单体, 而合成高分子往往是由重复的单体组成等. DNA分子的双股结构来源于各碱基层之间的π-π相互作用, 层与层之间的距离为3.4 Å.考虑到传统聚合方法的局限性和双股结构的特殊性, 有机化学家们经过不懈的努力, 精心设计骨架和连接基团的结构, 利用非共价键作用力或共价键作用力连接两条单股高分子, 形成双股的结构.因此, 本文将结合作者多年来在本领域的工作经验, 以连接基团之间的作用力为分类, 对人工合成类DNA双股高分子的历史及发展加以综述.
 表 1
生物高分子与合成高分子比较
Table 1.
Natural polymers vs. synthetic polymers
表 1
生物高分子与合成高分子比较
Table 1.
Natural polymers vs. synthetic polymers
生物高分子a 合成高分子 分子量分布(PDI) 1 >1 单体重复性 无 有 单体序列性 有 无 手性 有 有(依赖手性辅基) 双股螺旋性 有 十分困难 复制性 有 未知 催化性 有 稀少 a蛋白质、核酸等. 1 连接基团以非共价键相连接的双股高分子
在已知文献报道中, 连接基团以非共价键相连接, 构建双股高分子的结构主要由两种途径得以实现.第一, 单股高分子的自组装效应, 首先通过设计合适的单体制备出单股高分子, 然后单股高分子经由非共价键作用力(如π-π堆叠的相互作用、氢键、离子键、金属原子之间的吸引力等)通过自组装形成双股的结构.第二, 经由单体直接聚合, 单体自身即是由两侧的聚合单元和中间的连接基团所组成, 而这两部分以非共价键相连, 如离子键等, 在聚合过程中单体结构保持不变, 两侧分别独立地进行聚合从而得到双股高分子.在此类双股高分子中, 因为非共价键作用力往往较弱, 因此其双股的结构受外界因素影响较大, 如溶剂的选择、浓度、温度、pH值等.换言之, 只有在适当的外界条件下, 单股高分子的自组装过程才能顺利地进行, 得到双股高分子, 而该结构在外界因素改变时较难以保持.
1.1 自组装构建双股高分子
1994年, Kamachi等[13]首先合成出聚乙二醇的高分子链1, 链的两侧以萘基成酯作为末端基团, 之后将1与γ-环糊精2 (γ-CD, cyclodextrin)在水溶液中混合后, 可得到稳定的络合物3 (Eq. 1).研究发现, 当1和2(乙二醇单元:γ-环糊精)以2:1的比例混合时, 乙二醇单元被完全络合, 而此时环糊精单元的络合率仅50%.若此时额外加入更多的乙二醇高分子1, 可使环糊精单元几乎完全络合, 络合率达到90%, 该结果表明一个环糊精单元中包括四个乙二醇单元, 另一方面, 1H NMR的结果表明, 末端基团也同样与环糊精单元相络合, 若末端基团过大, 则无法形成稳定的络合物.在荧光光谱中, 观察到主要的激发波长位于400 nm, 来自于末端萘环二聚体(excimer)的激发, 而次要的激发波长位于340 nm, 对应于单个萘环的激发.这些结果证实络合物3为1和2通过自组装而成的双股结构, 其中两条高分子链穿过一个环糊精单元.
2001年, Lehn等[14]合成了一系列含有吡啶单元的寡聚物, 彼此之间以酰胺键相连.结果表明, 结构中含有7个吡啶单元的分子4在溶液中由于吡啶环之间的π-π相互作用及分子内的氢键, 构型会发生折叠, 形成单股螺旋结构.之后, 在以硝基苯为溶剂时, 随着分子4在溶液中浓度的增加, 1H NMR中δ 10.0附近出现新的信号峰且积分面积逐渐增加, 对应于自组装形成的二聚体5 (Scheme 2).此二聚体的X射线晶体衍射结果表明, 在空间纵向排列上, 吡啶环以面对面的方式通过范德华力进行堆叠, 平均距离为3.5 Å.增加溶液浓度后, 二聚体之间通过类似于单分子内的π-π堆叠和氢键进一步相互作用, 形成具有连续双股螺旋结构的高分子.
2007年, Sada等[15]则采用了另一种策略, 以刚性的连接基团6与柔性的两条单股高分子链7相连接, 在氢键的相互作用下形成类似于DNA分子的双股螺旋结构(Scheme 3).连接基团6由两端的手性噁唑啉配体(pybox)和中央的卟啉(porphyrin)单元所组成, 形成大面积共轭的刚性结构, 使得在形成双股高分子的骨架时, 层与层之间可以依靠π-π相互作用产生堆叠, 而由于噁唑啉单元手性的存在, 相邻两层之间的堆叠会扭曲一定的角度, 最终形成双股螺旋结构.单股高分子链7由聚丙胺的盐聚合而成, NH2+上的两个H与噁唑啉片段形成氢键, 构成双股高分子的结构.值得一提的是, 单股高分子链7单独存在时, 呈现无规则折叠状, 其双股的结构是由连接基团6诱导所形成的.在圆二色谱(CD, circular dichroism)测试中, 当没有任何添加时分子6在Soret带呈现正的Cotton效应, 来自于卟啉环和pybox配体之间的偶极耦合, 而当利用高分子7进行UV滴定时, Soret带上正的Cotton效应逐渐减少为负的效应, 证明卟啉环之间发生了扭曲和聚集, 以减弱结构中相邻异丙基所存在的空间位阻效应.利用原子力学显微镜(AFM, atomic force microscope)观察时发现, 高度2.3 nm对应于分子6的结构宽度, 而长度~86 nm则对应于单股高分子7的长度, 再次证明了该络合物双股的结构.
2006年至今, Yashima等[16]报道了一系列以胺和羧酸盐构成的盐桥为连接单元形成的双股高分子, 他们首先独立合成了单股高分子链8和9, 之后通过盐桥构建出具有互补结构的螺旋双股高分子10 (Scheme 4), 在后续的研究中也发现, 在手性胺络合的诱导作用下, 两条单股高分子链的螺旋方向会得到控制, 趋向于均一化.在圆二色谱测试中, 单独的高分子8呈现非常弱的Cotton效应, 而当与高分子9混合后, Cotton效应得到了极大的增强, 证明8与9通过盐桥发生了分子间的络合.该CD信号在混合后12 h逐渐增加, 证明双股络合物10的形成是一个消耗时间的过程, 进一步的实验表明, 在室温下以四氢呋喃为溶剂时, 整个络合过程的完成需要约36 h.动态光散射(DLS, dynamic light scattering)分析表明双股高分子10的平均流体动力学半径为(101±20) nm.
1990年, Kanatzidis等[17]首次报道了利用金原子之间的吸引力, 所形成的无机双股高分子11, 具有
$\left[{{\rm{AuS}}{{\rm{e}}_{\rm{5}}}} \right]_n^{n - }$ 的结构通式, 其中Au-Au原子的距离为2.95 Å.随后, 2004年Puddephatt等[18]合成了含有金原子的无机-有机单股高分子链12 (Scheme 5), 在X射线单晶衍射实验中观察到, 同样由于金原子之间的相互吸引力, 单股高分子12会发生自组装形成双股高分子13, 两条链之间Au-Au原子的距离为3.16 Å.与上述举例相类似, 2003年, Bosch等[19]利用四氟硼酸银与2, 3-二苯基吡嗪制备出含有银原子线性单股高分子14 (Scheme 6), 主链上银原子与吡嗪单元交替重复, 发现在选用硝基甲烷为溶剂时, 可以得到稳定的晶体.通过X射线晶体衍射结果表明, 两条单股高分子14以面对面, 吡嗪单元两两相对的形式, 通过自组装形成线型的双股高分子.这一结果可能是由于两条单股高分子链同时与溶剂分子硝基甲烷存在着相互作用, 使得两条单链相互靠近, 从而形成线性双股的结构, 而元素分析也表明银原子与配体吡嗪的比例为1:1.随后, Reger等[20]报道以六氟磷酸银和含有六个吡唑单元的化合物15, 制备出单股高分子16 (Scheme 6), 在溶液中同样可以通过自组装形成双股高分子, 驱动力则来源于单股高分子链间的π-π堆叠和C-H▪π相互作用.值得一提的是, 单股高分子16与14相比较, 由于重复单元构型上的不同, 其所形成的双股高分子空间上排列并非线型而是具有螺旋的结构.
1.2 单体直接聚合构建双股高分子
2001年, Matsumoto等[21]利用山梨酸和对苯二甲胺合成出晶体17作为单体, 随后在光照条件下进行自由基聚合反应, 两侧独立的进行聚合, 最终成功得到双股高分子18 (Eq. 2).双股高分子18的结构由X射线晶体衍射结果和红外光谱数据分析得到, 并进一步经由水解反应后所得到单股高分子的鉴定加以验证.此类双股高分子由于其刚性的结构, 往往溶解度较差, 因此表征和鉴定存在着一定的困难, 表征方法的选择就显得尤为重要.除此之外, 调整聚合方法, 控制双股高分子的分子量和聚合度以提高溶解性, 是另一个根本性的解决办法.
2 连接基团以共价键相连接的双股高分子
相较于上述以非共价键作用力连接构建双股高分子的实例, 以共价键作用力形成类似于DNA双股高分子的报道, 在以往的研究中非常少见.虽然以共价键作用力连接, 双股的结构将更加稳定, 层与层之间的距离也会得到更好的控制, 但该策略极大依赖于单体结构的设计, 聚合方法的选择, 溶解度的调控等因素, 因此具有很大的挑战性.在仅有的几例报道中, Anderson等[22]利用小角度中子散射(small angle neutron scattering)鉴定出以卟啉聚合物为骨架, 联吡啶单元为连接基团所形成的聚合物19为双股的结构(Eq. 3), 在近红外光谱(neat-IR)滴定实验中, 由于联吡啶的络合作用, 最终形成的双股高分子相较于络合前最大波长红移了75 nm. Zhang小组[23]则发展出由聚硅氧烷(polysiloxane)所构成的双股高分子, 首先利用单体分子间的氢键作用使分子排列成双股的结构, 之后再进行脱水聚缩合反应(polycondensation)从而得到双股高分子20和21 (Scheme 7).在红外光谱测量中, 单体于1107和1079 cm-1处的峰对应于Si—OEt, 聚合后则完全消失, 新出现的1077.15 cm-1对应于高分子20中Si—O—Si的振动.与之对应, 1H NMR也没有观察到Si—OEt所对应的峰, 证明单体已完全水解并聚合.通过差示扫描量热法(DSC, differential scanning calorimetry)测量, 得到双股高分子20的玻璃化转变温度为125.2 ℃.
2.1 降冰片烯骨架的发展
鉴于现有方法的局限性, 建立一种结构规整, 可适用于不同连接基团的高分子骨架, 是构建以共价键连接双股高分子领域亟待解决的问题.而近年来, 随着开环复分解聚合(ROMP, ring opening metathesis polymerization)的发展, 降冰片烯(norbornene)由于环张力的存在、结构的可修饰性和可控的空间构型等逐渐成为有机化学家们研究的热点.
20世纪50年代, Anderson和Merckling[24]首先开始研究具有环张力的烯烃在聚合反应中的表现.在80年代中期以前, 主要是采用含有Ti、V、W、Mo等过渡金属的Ziegler-Natta的催化体系[25]来催化反应的进行.例如, Montague等[26]于1960年将降冰片烯单体在TiCl4/ LiAl(C7H15)4/xylene的催化体系下进行聚合, 得到反式双键含量较高的高分子22, 其反应过程如Scheme 8所示.因其中间过渡态经历双键与金属原子配位的过程, 也被称为配位聚合(coordination polymerization).
 图式 8
降冰片烯的配位聚合
Scheme8.
Coordination polymerization of norbornene
图式 8
降冰片烯的配位聚合
Scheme8.
Coordination polymerization of norbornene
Rooney等[27]将催化体系换为ReCl5/C2H5AlCl2/ C2H5OH后, 作用于相同的降冰片烯单体时, 则可得到顺式cis双键含量较高的高分子.这类催化体系一般很难获得全反式或全顺式的高分子结构, 其中, 反式与顺式的结构大部分可以利用红外光谱来进行鉴别(trans≈965 cm-1, cis≈720 cm-1)[28], 而聚合物的立体规整度(tacticity)则主要是通过NMR来进行判断[27].这是由于重复单元中, 五元环上的碳容易受到周围环境的影响, 会在其13C NMR波谱中表现出差异.以Rooney等所得到的具有60%顺式结构的高分子为例, 因此聚合物并不是全顺式或全反式, 所以在双键的区域(δ 133~135)显得比较复杂.另外, 也可以看出, 在五元环重复单元中, C(1)和C(4)受到旁边顺式反式双键的影响, 会有顺式顺式(cc)、顺式反式(ct)、反式顺式(tc)和反式反式(tt)四种不同的信号峰, 这一结果说明若降冰片烯聚合物同时有顺式和反式的结构, 其鉴定会比较困难.
1990年, Schrock[29]发展出一类新型的催化剂, 在催化剂中过渡金属原子与碳原子直接以双键相连, 称之为卡宾.如钼卡宾和钨卡宾催化剂(Schrock催化剂), 这是第一个能应用于各种开环复分解反应的催化剂, 具有很高的反应活性; 1995年, Grubbs等[30, 31]合成出一系列钌卡宾催化剂(Grubbs-Ⅰ催化剂), 是一种对于开环复分解聚合反应(ROMP)十分有效的催化剂.较前者而言, 其在空气中具有一定的稳定性且反应速率快.因此, 它被广泛应用于有机反应, 材料化学以及天然产物的合成中.随后, Herrmann和Grubbs等[32]通过改变不同的配位基团, 合成出反应性更高的第二代的钌卡宾催化剂(Grubbs-Ⅱ催化剂).它们的结构如Scheme 9所示.
 图式 9
Schrock, Grubbs-Ⅰ, Grubbs-Ⅱ催化剂结构
Scheme9.
Structures of Schrock, Grubbs-Ⅰ, Grubbs-Ⅱ catalysts
图式 9
Schrock, Grubbs-Ⅰ, Grubbs-Ⅱ催化剂结构
Scheme9.
Structures of Schrock, Grubbs-Ⅰ, Grubbs-Ⅱ catalysts
1994年, Schrock等[33]使用钼卡宾催化剂催化聚合具有光学活性的手性降冰片二烯单体(norbornenediene) 23, 并利用二维NMR谱图(COSY, correlation spectroscopy)解析出具有光学活性的聚合物24和25 (Scheme 10).其中, 因为24的立体结构为间同排列, 在双键上有一个对称轴C2的存在, 五元环上的氢原子化学环境相同(Ha), 所以在二维波谱中两个Ha不会彼此耦合.而不具备此对称性的全同立体排列高分子25的五元环上氢原子化学环境并不相同(Ha, Hb), 所以在二维波谱中可以看到两个不同化学环境的氢互相耦合.
Noels小组[34]报道了利用钌金属催化剂对单体26进行聚合后得到高分子27 (Scheme 11), 首先通过1H NMR中双键化学位移仅在δ 5.41显示为单峰, 确定双键为全反式的构型.之后在13C NMR中观察到降冰片烯上七个碳原子中的六个都给出单峰, 如C(7)在δ 39.3处为单峰.结合Rooney[27]的结果, 若有交叉的构型存在, 如顺式反式(tc)或反式反式(cc), 则C(7)的位置分别是δ 40.0和40.8;而这些信号峰并不存在, 说明高分子27的双键为全反式构型.值得注意的是, 在δ 45.3和45.4 [C(1), C(4)]处有两个峰互相重叠, 而主要的峰位于更高场的位置, 说明高分子27的结构大部分为全同的排列.从13C NMR波谱中可以清楚的看到其还原产物28在C(1), C(4)的位置上简化为两个峰.主要的信号峰为m, 代表内消旋(meso), 较低的信号峰为r代表着外消旋(racemic).因此, 可以确定高分子27主要为反式全同的排列方式.
1997年, Grubbs等[35]以降冰片烯或环丁烯为骨架, 引入具有液晶性质的基团作为悬挂基团, 以Grubbs-Ⅰ催化剂催化开环复分解聚合反应制备出高分子29和30 (Scheme 12), 它们都表现出液晶的性质.偏光显微照片显示, 高分子29为向列型液晶(nematic liquid crystal), 而30为近晶型液晶(sematic liquid crystal).这可能是由于高分子30中悬挂基团排列比较有序, 才会使其具有近晶型液晶的性质.
为了进一步精确控制降冰片烯的空间构型, 使之可以作为构建类DNA双股高分子的骨架, 在上述工作基础上, 台湾大学Luh小组设计和合成一类新型的降冰片烯骨架, 并对其空间结构和立体规整度作了详尽而系统的研究. 1999年, 他们报道了一系列降冰片烯衍生物经钌卡宾(Grubbs-Ⅰ)催化开环复分解聚合反应[36]所得的聚合物31, 分子量从4200到17000.在超瑞利散射(HRS, hyper-Rayleigh scattering)的实验中观察到在此分子量范围内, 非线性光学性质(β0)随着高分子分子量的增加而增加(Scheme 13), 表明其立体化学基本上是一致的.同时良好的线性关系(R2=0.97), 表明发色团的方向在空间排列上具有一致性, 均大致朝一个方向同向syn排列, 因此这类高分子呈梳子状且具有一定的刚性, 而该直线的斜率(slope)则对应于相邻两降冰片烯单元之间夹角的正切值tgθ.
继而, Luh小组[37]于2006年合成了刚性较大的的高分子32 (Scheme 14), 以原子力学显微镜(AFM)观察, 发现这类高分子具有柱状的形态, 这充分说明了该高分子是一刚性聚合物.而后, 利用轻敲式原子力学显微镜(tapping mode AFM)对以卟啉为悬挂基团的单股高分子33进行观察时[38], 发现聚合物33可以在HOPG (highly ordered pyrolytic graphite)表面自组装成平行的条纹形状, 这些条纹之间的间距约为5 nm.
Luh小组[38]又对比以酯基连接的含卟啉悬挂基团的降冰片烯单体34和其对应的高分子35的1H NMR波谱(Scheme 15), 相较于单体而言, 高分子卟啉环上β-氢受到邻近的卟啉结构强烈的屏蔽效应, 讯号明显向高场位移.同时由紫外吸收光谱比较可以看出, 单体34中卟啉环的Q-bands吸收峰在553及593 nm, 在高分子35中卟啉的Q-bands红移至558及600 nm.此外, 单体34的Soret band吸收在420 nm, 而高分子35中卟啉的Soret band吸收分裂为两个吸收峰, 分别位于406及421 nm, 表明相邻的卟啉发色团之间有明显的激发子耦合现象(excitation coupling).上述实验结果表明, 悬挂在高分子骨架上相邻的卟啉基团具有规律性的排列且朝向同一个方向.
在利用钌卡宾得到一系列N-芳基取代的5, 6-endo吡咯并环的降冰片烯的单股聚合物36 (Eq. 4)后[39], 比较它们的紫外吸收光谱, 聚合物的最大吸收波长相对于单体都有蓝移的现象, 消光系数则逐渐减少, 这是由于降冰片烯单体具有一定的环张力, 随着开环复分解聚合反应的进行则会释放部分的张力.
更重要的是, Luh小组[39]通过非线性光学和13C NMR波谱研究了单股高分子36的立体规整度(tacticity) (Scheme 16).一般而言, 降冰片烯聚合物的取代基位向所形成的构型(configuration)共有三种不同的排列方式, 分别为全同立体排列(isotactic)、间同立体排列(syndiotactic)和不规则排列(atactic).而由之前的研究可知, Luh小组所发展出的降冰片烯骨架为同向syn整齐排列, 所以在不考虑anti位向的情形下, 其可能有四种不同的空间排列, 如图式 16所示.在反式间同排列(trans syndiotactic)和顺式全同排列(cis isotactic)的情形下, 相邻两个降冰片烯单元之间存在着C2的对称轴, 而在反式全同排列(trans isotactic)和顺式间同排列(trans syndiotactic)的情形下, 相邻两个降冰片烯单元之间则无任何对称元素.采用Grubbs-Ⅰ作为催化剂得到高分子36时, 由1H NMR (δ 5.36)和IR (964 cm-1)可判断双键的构型为反式(trans), 而当高分子36中取代基为吸电子基团如CO2Et(36b), C(7)在δ 36附近给出双重峰(doublet), 在双键还原后又变为单峰(singlet), 这就说明在36b中相邻两个降冰片烯单元之间并无对称元素, 结合Scheme 16可以确定该类降冰片烯单股聚合物的立体规整度为反式全同排列(trans isotactic).综合以上研究, 可以确定这类N-芳基取代的5, 6-endo 吡咯并环的降冰片烯单股聚合物为取代基同向排列syn的刚性结构, 且为具有反式双键的全同立体排列(trans isotactic), 可作为构建双股高分子的骨架.
2.2 以降冰片烯为骨架构建类DNA双股高分子的发展
结合降冰片烯单股高分子的结构特点, 如果在单股降冰片烯悬挂基团的末端引入另一个降冰片烯单元(bisnorbornene), 如单体37所示, 在开环复分解聚合反应时, 则可能得到三种形式的高分子38、40和42[40] (Scheme 17).如果单体37的连接基团未经特别设计和安排, 则经过聚合反应后很大可能会得到交联(cross linking)高分子38.值得注意的是, 在这种情形下, 高分子38将拥有两组以上的末端基团(red and blue ball).而若双环化烯烃单体37以刚性基团相连接, 由于连接基团之间和降冰片烯骨架之间的相互作用力, 进行开环复分解聚合后则将会得到类似于40结构的双股高分子.在这种情况下, 单体37在聚合反应中经过有序的反应过程, 通过类似于39的过渡态, 得到含有两组末端基团的梯蕃聚合物40.第三种可能性则是若连接基团具有一定的柔软性, 则有可能首先在单体37分子内部发生环化复分解反应, 产生一个新的卡宾中间体41, 再与另一分子的双环化烯烃单体37反应, 不断发生聚合进而得到环化单股高分子42.这样所得到的单股高分子在高分子链的末端只会有一组连接基团.由此可知, 连接基团的选择对于双股高分子结构的构建是至关重要的.
在上述单股高分子工作的基础之上, Luh小组[41]综合考虑空间距离等因素, 最终选用二茂铁为连接基团, 首次成功合成出类似于DNA的双股高分子43 (Eq. 5), 从而也实现了Scheme 17中所提及的第二种情况.通过单晶衍射图像分析可以得出, 单股高分子链中相邻两个降冰片烯单元之间的距离为5~6.5 Å, 而二取代的二茂铁的单晶中, 相邻铁原子之间的距离为5.5~5.9 Å[42], 所以其可以很好的排列于降冰片烯骨架之间而不会有太大阻碍.在利用扫描隧道显微镜(STM, scanning tunneling microscope)观察以二茂铁为连接基团的双股高分子43[41, 43, 44]时可清楚的观察到三种不同的双股构型, 分别为螺旋状(helix)、大螺旋状(supercoil)和梯状(ladder), 并且从图上可以知道其整个分子宽度为22 Å左右, 而重复单元的距离约为4.5~5 Å.
将双股高分子43进行醇解反应, 得到相对应的二茂铁单元44和单股聚合物45 (Scheme 18), 单体46同样以钌卡宾催化剂进行开环复分解聚合反应, 得到的单股聚合物与45的波谱性质完全一样, 以上的结果充分显示43确为双股聚合物, 这是首次以开环复分解聚合反应得到双股聚合物, 也是首次鉴定了这类双股聚合物的结构, 而所采用的这一策略也可以用于鉴定双股及相关高分子的结构.
2007年, Luh小组[45] (Scheme 19)将降冰片烯酸的酰氯接至具有良好立体化学规整度的单股高分子47, 进行酯化反应得到含有二茂铁连接基团的不对称双降冰片烯分子骨架单股高分子48, 之后再进行开环复分解聚合反应, 得到双股高分子49, 利用扫描隧道显微镜观察到其具有类似于双股高分子43的刚性梯形结构.若将49在碱性条件下水解, 得到相对应的水溶性高分子后, 经盐酸酸化可以转变为中性的羧酸高分子50.整个过程如同DNA利用互补碱基进行复制, 将信息从模板高分子传递到相对应互补的高分子.这一实验结果不仅第一次实现了人工合成双股高分子的复制, 也为非对称(unsymmetrical)双股高分子的合成提供了方法.
在研究双股高分子49在HOPG表面自组装情况时, Luh小组[46]发现将49溶解在辛苯(phenyloctane)中配制成较高浓度溶液(>300 nmol/L)时, 其可在HOPG表面形成很好的二维结构.因为单个高分子链的平均长度为10~15 nm, 所以各高分子链间依靠末端基团(PhCH=和=CH2)的π-π相互作用在主链方向上聚集, 从而形成大范围的梯形二维结构, 如Scheme 20所示.
在成功制备出以二茂铁为连接基团的双股高分子后, Luh小组[47]继续以聚降冰片烯衍生物为骨架, 通过扩展中间的连接基团, 将二茂铁改变成蒽、低聚噻吩、卟啉、立方烷等刚性基团, 合成了一系列“梯形”(ladder)的双股高分子51a~51n, 并将拥有这类结构的高分子命名为“梯蕃高分子”(polymeric ladderphane)[48] (Scheme 21).梯蕃高分子为一梯状高分子, 是以多层连接基团及双股或多股高分子骨架经由共价键的连接方式所组成, 因此梯蕃高分子的连接基团与骨架是垂直的, 并且可以是二维的平面芳香环、大环金属络合物或有机金属衍生物.梯蕃高分子也可被视为以高分子骨架来连接的多层环蕃(cyclophanes), 或更确切地说, 梯蕃高分子是以高分子骨架为系链的直线且共面排列(cofacially aligned)的芳香烃(arenes).
利用扫描隧道显微镜(STM)观察这类双股高分子时, 也同样可以看到与43和49相类似, 在HOPG表明自组装聚集而成的二维梯形结构, 这类高分子也同样借由末端基团的π-π相互作用及高分子股架之间的范德华力, 在HOPG表面上以规则的方式堆叠排列.同时也观察到, 在双股高分子链中, 发光团排列整齐, 这样的排列方式使得发光团之间具有相互作用力, 从而影响发光团的光物理性质.
由这些实例可以看出, Luh小组首先所发展出N-芳基取代的5, 6-endo吡咯并环的降冰片烯衍生物为骨架, 在引入刚性连接基团后, 利用Grubbs-Ⅰ催化剂催化开环复分解聚合反应, 成功得到反式(E)全同立体排列的双股梯蕃高分子.刚性的连接基团是构建双股高分子结构的关键, 因为足够的刚性才能使单体两侧的两个双键远离, 从而两边可以独立的进行ROMP反应.另外, 由于连接基团的π-π相互作用, 使得单体靠近时排列整齐, 同侧的降冰片烯反应速率较快, 所以在聚合过程中也不会发生随机的交联反应(random cross linking).这些因素都保证了单体在聚合过程中可以依Scheme 17中第二种情况所示, 最终得到排列整齐的双股梯蕃高分子.
而与之相对, 在烯烃复分解中选择性的得到全顺式(Z)双键构型的反应, 一直以来也是化学家们所研究的热点[49~53].其中比较有代表性的是Schrock所发展出的钼(Mo)卡宾或者钨卡宾(W)催化剂可以通过配体的改变, 在各类烯烃复分解反应诸如自偶联反应(homocoupling)[50]、交叉复分解反应(CM, cross metathesis)[51]、环化复分解反应(RCM, ring closing metathesis)[52]和开环聚合复分解反应(ROMP, ring opening metathesis polymerization)[53]中都表现出很高的顺式选择性(Z-selectivity).
在制备高分子中, Schrock等[53a]利用钼卡宾催化剂53催化降冰片二烯的单体52进行开环复分解聚合反应, 最终得到了>99%顺式(Z)的间同排列的高分子(Eq. 6), 如Scheme 16中cis syndiotactic所示意.
而后, Schrock等[53c]采用与53相类似的催化剂54催化降冰片烯单体聚合时, 发现当采用消旋(racemic)的单体55 (endo, exo-5, 6-dicarbomethynorbornene, DCM-NBE)时, 会得到顺式间同排列的高分子(Eq. 7), 而当采用单一构型的单体时, 如(+)-55, 以同样的催化剂54催化, 则会得到同时含有全同排列(isotactic)和间同排列(syndiotactic)结构的混合体系.
这是因为当采用(rac)-55时, 由于单体分子较小, 其有可能会从a, b两个方向靠近钼卡宾(Scheme 22).反应起始阶段, 当一种构型的单体, 如(-)-55从a方向靠近, 形成金属四元环过渡态Ⅰ, 经过开环复分解后得到中间体Ⅱ, 可以看出Ⅱ中钼卡宾双键的位相发生了改变, 由偏向外侧改为偏向里侧.这时, 另一种构型的单体(+)-55, 从b方向靠近钼卡宾形成金属四元环过渡态 Ⅲ.经开环复分解后, 得到含有交替五元环片段的中间体Ⅳ, 此时钼卡宾双键的位相再次发生改变, 由先前Ⅱ中的偏向里侧变为偏向外侧, (-)-55单体继续从a方向进攻钼卡宾形成Ⅴ中间体, 开环复分解后Ⅵ中钼卡宾的双键偏向里侧, 此时又有利于(+)-55从b方向进攻.总结规律可以看出, (-)-55从a方向进攻后, 双键位相发生改变, 有利于(+)-55从b方向进攻.之后, 双键位相再次改变, 又有利于(-)-55从a方向进攻, 如此循环往复, 所得到的五元环构型呈交替排列, 最终得到顺式间同(cis syndiotactic)排列的高分子.
 图式 22
顺式间同排列聚降冰片烯形成机理示意图
Scheme22.
Mechanism for the formation of cis syndiotactic poly-norbornene
图式 22
顺式间同排列聚降冰片烯形成机理示意图
Scheme22.
Mechanism for the formation of cis syndiotactic poly-norbornene
进一步研究中, Schrock等[53c]发现若在钼卡宾催化剂中引入更大的位阻基团, 例如含多取代的联苯类配体的催化剂56b, 由于联苯基团空间上的分布极大的阻挡了从其中一个方向进攻的可能, 再加之使用光学活性的(+)-55为原料, 成功实现了单体只从单一的方向上靠近卡宾, 从而制备得到顺式全同排列的高分子产物(Eq. 8).
2012年, Luh小组[54]与Schrock小组合作, 将钼卡宾催化剂56a应用于所发展的N-芳基取代的5, 6-endo吡咯并环的降冰片烯单体57, 制备出顺式全同排列的单股高分子58 (Eq. 9).在该高分子的13C NMR波谱中, δ 39.2, 40.4, 47.5和49.4的峰分别对应于单体中降冰片烯单元的部分, 而它们均为单峰说明在高分子链中相邻两个降冰片烯单体中存在对称面, 则高分子中双键的构型应为顺式(cis configuration).与之相反, 相同的单体57以Grubbs-Ⅰ催化剂聚合所得反式(trans)全同排列的单股高分子36b中, 由于相邻的单体单元之间没有对称元素, C(7)在δ 36附近显示为双重峰.这一结果是由于单体57在endo位置上的位阻比56要大的多, 为了避免N-Aryl基团与Mo=NAr之间的位阻, 降冰片烯单体将更倾向于通过旋转, 与起始卡宾单元反应, 因此所有的单体将会从同一方向靠近钼卡宾, 最终得到顺式全同(cis isotactic)排列构型的高分子.
在此基础上, Luh小组[54]以二茂铁为连接基团, 通过钼卡宾催化剂56a, 首次成功制备出以降冰片烯为骨架顺式全同排列的双股梯蕃高分子59 (Scheme 23), 并通过水解反应和与反式全同排列高分子的比较, 证明了其立体规整度.随后在STM实验发现双股高分子59也同样可以在HOPG表面聚集成有序的二维结构呈现带状排列, 每一带的宽度~2.5 nm, 相邻单体间的间距为0.5~0.6 nm, 与高分子的骨架结构相对应.但是由于末端基团的不同, 在单体结构相同的情况下, 其稳定性比反式全同排列的双股高分子43稍弱.
3 结论与展望
综上所述, 人工合成类似于DNA的双股高分子, 并模拟DNA分子的复制过程是一个重要的研究领域.近年来, 随着自组装化学及有机合成技术的发展, 化学家们将连接基团以非共价键或共价键相连, 成功构建出一系列结构为双股的梯形高分子.当连接基团以非共价键连接时, 在π-π堆叠的相互作用、氢键、离子键、金属原子之间的吸引力等作用力下, 单体经由自组装形成双股的结构, 其结构的鉴定多采用间接的方式, 如核磁共振波谱、圆二色谱、紫外吸收光谱、荧光光谱等, 但是此类双股高分子的结构稳定性不足, 且容易受外界因素如温度, 浓度等的影响.当连接基团以共价键相连时, 受限于单体的结构设计, 连接基团的空间排列等, 所报道的例子虽仍有局限性, 但是聚降冰片烯被证明是一类有效的高分子骨架, 可适用于各种不同的刚性连接基团, 成功构建了一系列立体规整的双股高分子, 具有优异的普适性.此类以平面芳香环、大环金属络合物或有机金属衍生物等为连接基团, 两侧骨架直线排列所形成的双股高分子被命名为梯蕃高分子, 其结构的鉴定可采用直接的方式, 如扫描隧道显微镜等.
纵观类DNA双股高分子的合成方法及策略, 需要综合考虑以下几个重要因素:一、骨架结构的选择应具有一定的刚性, 使高分子能在空间上排列规整有序; 二、取代基的存在会改变双股高分子的折叠形态和手性, 但过大的空间位阻也会阻碍高分子的形成; 三、聚合方法应以活性聚合为主, 可精确控制与调整高分子的分子量和分子量分布.在今后的研究中, 开发新的高分子骨架, 与功能性连接基团相结合, 进一步拓展双股高分子的种类, 将是未来需要研究的重点课题之一.此类双股高分子由于具有结构的独特性, 与传统高分子相比性质上有很大差异, 在分子机器、新型光电材料、仿生材料等领域具有十分重要的潜在应用性.
-
-
[1]
Watson, J. D.; Crick, F. H. C. Nature 1953, 171, 737. doi: 10.1038/171737a0
-
[2]
(a) Schuster, G. B. Acc. Chem. Res. 2000, 33, 253.
(b) Fink, H.-W.; Schonenberger, C. Nature 1999, 398, 407.
(c) Abi, A.; Ferapontova, E. E. J. Am. Chem. Soc. 2012, 134, 14499.
(d) Genereux, J. C.; Boal, A. K.; Barton, J. K. J. Am. Chem. Soc. 2010, 132, 891.
(e) Duprey, J.-L. H. A.; Carr, S. J.; Horswell, S. L.; Kowalski, J.; Tucker, J. H. R. J. Am. Chem. Soc. 2015, 138, 746. -
[3]
(a) Gill, R.; Zayats, M.; Willner, I. Angew. Chem., Int. Ed. 2008, 47, 7602.
(b) Cheng, C. S.; Rai, K.; Garber, M.; Hollinger, A.; Robbins, D.; Anderson, S.; Macbeth, A.; Tzou, A.; Carneiro, M. O.; Raychowdhury, R.; Russ, C.; Hacohen, N.; Gershenwald, J. E.; Lennon, N.; Nusbaum, C.; Chin, L.; Regev, A.; Amit, I. Nat. Commun. 2013, 4, 3672.
(c) Houlton, A.; Pike, A. R.; Galindo, M. A.; Horrocks, B. R. Chem. Commun. 2009, 14, 1797. -
[4]
(a) Kasumov, A. Y.; Kociak, M.; Gueron, S.; Reulet, B.; Volkov, V. T.; Klinov, D. V.; Bouchait, H. Science 2001, 291, 280.
(b) Hopkins, D. S.; Pekker, D.; Goldbart, P. M.; Bezryadin, A. Science 2005, 308, 1762.
(c) Watson, S. M. D.; Pike, A. R.; Pate, J.; Houlton, A.; Horrocks, B. R. Nanoscale 2014, 6, 4027. -
[5]
(a) Joyce, G. F. Nature 1989, 338, 217.
(b) Bochman, M. L.; Schwacha, A. Nature 2015, 524, 186.
(c) Dewar, J. M.; Budzowska, M.; Walter, J. C. Nature 2015, 525, 345.
(d) Creager, R. L.; Li, Y.-L.; Macalpine, D. M. Cell 2015, 161, 418.
(e) Bell, S. P. Science 2014, 346, 418. -
[6]
Orgel, L. E. Nature 1992, 358, 203. doi: 10.1038/358203a0
-
[7]
(a) Von Kiedrowski, G. Angew. Chem., Int. Ed. 1986, 25, 932.
(b) Brandsch, R.; Luther, A.; Von Kiedrowski, G. Nature 1998, 396, 245.
(c) Bag, B. G.; Von Kiedrowski, G. Pure Appl. Chem. 1996, 68, 214. -
[8]
Peins, L. J.; Reinhoudt, D. N.; Timmerman, P. Angew. Chem., Int. Ed. 2001, 40, 2382. doi: 10.1002/(ISSN)1521-3773
-
[9]
(a) Tjivikua, T.; Ballester, P.; Rebek, J. Jr. J. Am. Chem. Soc. 1990, 112, 1249.
(b) Feng, Q.; Park, P. K.; Rebek, J., Jr. Science 1992, 256, 1179.
(c) Conn, M. M.; Winter, E. A.; Rebek, J. Jr. Acc. Chem. Res. 1994, 27, 198. -
[10]
Würthner, F.; Rebek, J.Angew. Chem., Int. Ed. 1995, 34, 446. doi: 10.1002/(ISSN)1521-3773
-
[11]
Terfort, A.; Von Kiedrowski, G. Angew. Chem., Int. Ed. 1992, 31, 654. doi: 10.1002/(ISSN)1521-3773
-
[12]
Wang, B.; Sutherland, I. O. Chem. Commum. 1997, 1495. http://d.wanfangdata.com.cn/Periodical/jsjxb201411015
-
[13]
Harada, A.; Li, J.; Kamachi, M. Nature 1994, 370, 126. doi: 10.1038/370126a0
-
[14]
(a) Berl, V.; Huc, I.; Khoury, R. G.; Lehn, J.-M. Chem.-Eur. J. 2001, 7, 2810.
(b) Berl, V.; Huc, I.; Khoruy, R. G.; Lehn, J.-M. Chem.-Eur. J. 2001, 7, 2789. -
[15]
Sugimoto, T.; Suzuki, T.; Shinkai, S.; Sada, K. J. Am. Chem. Soc. 2007, 129, 270. doi: 10.1021/ja067613h
-
[16]
(a) Ikeda, M.; Tanaka, Y.; Hasegawa, T.; Furusho, Y.; Yashima, E. J. Am. Chem. Soc. 2006, 128, 6806.
(b) Maeda, T.; Furusho, Y.; Sakurai, S.-I.; Kumaki, J.; Okoshi, K.; Yashima, E. J. Am. Chem. Soc. 2008, 130, 7938.
(c) Makiguchi, W.; Kobayashi, S.; Furusho, Y.; Yashima, E. Angew. Chem., Int. Ed. 2013, 52, 5275. -
[17]
Park, Y.; Kanatzidis, M. G. Angew. Chem., Int. Ed. 1990, 29, 914. doi: 10.1002/(ISSN)1521-3773
-
[18]
Mohr, F.; Jennings, M. C.; Puddephatt, R. J. Angew. Chem., Int. Ed. 2004, 43, 969. doi: 10.1002/(ISSN)1521-3773
-
[19]
Schultheiss, N.; Powell, D. R.; Bosch, E. Inorg. Chem. 2003, 42, 8886. doi: 10.1021/ic034994g
-
[20]
Reger, D. L.; Semeniuc, R. F.; Rassolov, V.; Smith, M. D. Inorg. Chem. 2004, 43, 537. doi: 10.1021/ic035207i
-
[21]
Nagahama, S.; Matsumoto, A. J. Am. Chem. Soc. 2001, 123, 12176. doi: 10.1021/ja011575e
-
[22]
Screen, T. E. O.; Thorne, J. R. G.; Denning, R. G.; Bucknall, D. G.; Anderson, H. L. J. Am. Chem. Soc. 2002, 124, 9712. doi: 10.1021/ja026205k
-
[23]
Tang, H.; Sun, J.; Jiang, J.; Zhou, X.; Hu, T.; Xie, P.; Zhang, R. J. Am. Chem. Soc. 2002, 124, 10482. doi: 10.1021/ja025650c
-
[24]
Anderson, A. W.; Merckling, N. G. US 2721189, 1955[Chem. Abstr. 1956, 50, 3008i].
-
[25]
(a) Ziegler, K.; Holzkamp, E.; Breil, H.; Martin, H. Angew. Chem. 1955, 67, 426.
(b) Natta, G. J. Polym. Sci. 1955, 16, 143. -
[26]
Truett, W. L.; Johnson, D. R.; Robinson, I. M.; Montague, B. A. J. Am. Chem. Soc. 1960, 82, 2337. doi: 10.1021/ja01494a057
-
[27]
(a) Ivin, K. J.; Laverty, D. T.; Rooney, J. J. Makromol. Chem. 1977, 178, 1545.
(b) Ivin, K. J.; Laverty, D. T.; Rooney, J. J.; Watt, P. Recl. Trav. Chim. Pays-Bas 1977, 96, 54.
(c) Ivin, K. J.; Lapienis, G.; Rooney, J. J. Chem. Commun. 1979, 1068.
(d) Ivin, K. J.; Lapienis, G.; Rooney, J. J. Polymer 1980, 21, 436. -
[28]
Beaven, G. H.; Johnston, E. A.; Miller, R. G.; Willis, H. A. Molecular Spectroscopy, Heywood & Co., Ltd., London, 1961. http://d.wanfangdata.com.cn/Periodical/jsjxb201411015
-
[29]
(a) Schrock, R. R. Acc. Chem. Res. 1990, 23, 158.
(b) Schrock, R. R.; Murdzek, J. S.; Bazan, G. C.; Robbins, J.; Dimare, M.; Oregan, M. J. Am. Chem. Soc. 1990, 112, 3875.
(c) Schrock, R. R.; Hoveyda, H. Angew. Chem., Int. Ed. 2003, 42, 4592. -
[30]
(a) Kanaoka, S.; Grubbs, R. H. Macromolecules 1995, 28, 4707.
(b) Schwab, P.; France, M. B.; Ziller, J. W.; Grubbs, R. H. Angew. Chem., Int. Ed. 1995, 34, 2039.
(c) Schwab, P.; Grubbs. R. H. J. Am. Chem. Soc. 1996, 118, 100.
(d) Weck, M.; Schwab, P.; Grubbs, R. H. Macromolecules 1996, 29, 1789.
(e) Trnka, T. M.; Grubbs, R. H. Acc. Chem. Res. 2001, 34, 18. -
[31]
(a) Grubbs, R. H.; Novak, B. M.; McGrath, D. M.; Benedicto, A.; France, M.; Nguyen, S. T. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.)1992, 203, 20.
(b) Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100.
(c) Nguyen, S. T.; Grubbs, R. H. J. Am. Chem. Soc. 1992, 114, 3974. -
[32]
(a) Weskamp, T.; Schattenmann, W. C.; Spiegler, M.; Herrmann, W. A. Angew. Chem., Int. Ed. 1998, 37, 2490.
(b) Ackerman, L.; Fürstner, A.; Weskamp, T.; Kohl, F. J.; Herrmann, W. A. Tetrahedron Lett. 1999, 40, 4787.
(c) Scholl, M.; Trnka, T. M.; Morgan, J. P.; Grubbs, R. H. Tetrahydron Lett. 1999, 40, 2247. -
[33]
O'Dell, R.; McConville, D. H.; Hofmeister, G. E.; Schrock, R. R. J. Am. Chem. Soc. 1994, 116, 3414. doi: 10.1021/ja00087a028
-
[34]
Delaude, L.; Demonceau, A.; Noels, A. F. Macromolecules 2003, 36, 1446. doi: 10.1021/ma021315x
-
[35]
(a) Maughon, B. R.; Weck, M.; Mohr, B.; Grubbs, R. H. Macromolecules 1997, 30, 257.
(b) Weck, M.; Mohr, B.; Maughon, B. R.; Grubbs, R. H. Macromolecules 1997, 30, 6430. -
[36]
(a) Sattigeri, J. A. ; Shiau, C. -W. ; Yeh, F. F. ; Jin, B. -Y. ; Hsu, C. C. ; Luh, T. -Y. J. Am. Chem. Soc. 1999, 121, 1607.
(b) Shiau, C. -W. ; Sattigeri, J. A. ; Shen, C. K. -F. ; Luh, T. -Y. Organometallics 1999, 572, 291.
(c) Hsu, C. C. ; Hung, T. -H. ; Liu, S. ; Yeh, F. -F. ; Jin. B. -Y. ; Sattigeri, J. A. ; Shiau, C. -W. ; Luh, T. -Y. Chem. Phys. Lett. 1999, 311, 355.
(d) Churikov, V. M. ; Hung, M. -F. ; Hsu, C. C. ; Shiau, C. -W. ; Luh, T. -Y. Chem. Phys. Lett. 2000, 332, 19.
(e) Luh, T. -Y. ; Chen, R. -M. ; Hung, T. -Y. ; Basu, S. ; Shiau, C. -W. ; Lin, W. -Y. ; Jin, B. -Y. ; Hsu, C. C. Pure Appl. Chem. 2001, 73, 243.
(f) Xiao, C. -W. M. S. Thesis, National Taiwan University, Taipei, 1999 (in Chinese).
(萧崇玮, 硕士论文, 台湾大学, 台北, 1999. ) -
[37]
Lin, W.-Y.; Murugesh, M. G.; Sudhakar, S.; Yang, H.-C.; Tai, H.-C.; Chang, C.-S.; Liu, Y.-H.; Wang, Y.; Chen, I.-W. P.; Chen, C.-H.; Luh, T.-Y. Chem.-Eur. J. 2006, 12, 324. doi: 10.1002/(ISSN)1521-3765
-
[38]
Wang, H.-W.; Liu, Z.-C.; Chen, C.-H.; Lim, T.-S.; Fang, WS.; Chao, C.-G.; Yu, J.-Y.; Lee, S.-L.; Chen, C.-H.; Huang, S.-L.; Luh, T.-Y. Chem.-Eur. J. 2009, 15, 5719. doi: 10.1002/chem.v15:23
-
[39]
Lin, W.-Y.; Wang, H.-W.; Liu, Z.-C.; Xu, J.; Chen, C.-W.; Yang, Y.-C.; Huang, S.-L.; Yang, H.-C.; Luh, T.-Y. Chem. Asian J. 2007, 2, 764. doi: 10.1002/(ISSN)1861-471X
-
[40]
Zhu, L.; Lin, N.-T.; Xie, Z.-Y.; Lee, S.-L.; Huang, S.-L.; Yang, J.-H.; Lee, Y.-D.; Chen, C.-H.; Chen, C.-H.; Luh, T.-Y. Macromolecules 2013, 46, 656. doi: 10.1021/ma302293q
-
[41]
Yang, H.-C.; Lin, S.-Y.; Yang, H.-C.; Lin, C.-L.; Tsai, L.; Huang, S.-L.; Chen, I.-W. P.; Chen, C.-H.; Jin, B.-Y.; Luh, T.-Y. Angew. Chem., Int. Ed. 2006, 45, 726. doi: 10.1002/(ISSN)1521-3773
-
[42]
Brock, C. P.; Fu, Y. Acta Crystallogr., Sect B: Struct. Sci. 1997, 53, 928. doi: 10.1107/S0108768197005132
-
[43]
(蔡纶, 硕士论文, 台湾大学, 台北, 2002. )Cai, L. M.S. Thesis, National Taiwan University, Taipei, 2002 (in Chinese).
-
[44]
(杨慧君, 博士论文, 国立台湾大学, 台北, 2006. )Yang H.-J. Ph.D. Dissertation, National Taiwan University, Taipei, 2006 (in Chinese).
-
[45]
Lin, N.-T.; Lin, S.-Y.; Lee, S.-L.; Chen, C.-H.; Hsu, C.-H.; Hwang, L.-P.; Xie, Z.-Y.; Chen, C.-H.; Huang, S.-L.; Luh, T.-Y. Angew. Chem., Int. Ed. 2007, 46, 4481. doi: 10.1002/(ISSN)1521-3773
-
[46]
Lee, S.-L.; Lin, N.-T.; Liao, W.-C.; Chen, C.-H.; Yang, H.-C.; Luh, T.-Y. Chem. Eur. J. 2009, 15, 11594. doi: 10.1002/chem.200901634
-
[47]
(a) Chou, C.-M.; Lee, S.-L.; Chen, C.-H.; Biju, A. T.; Wang, H.-W.; Wu, Y.-L.; Zhang, G.-F.; Yang, K.-W.; Lim, T.-S.; Huang, M.-J.; Tsai, P.-Y.; Lin, K.-C.; Huang, S.-L.; Chen, C.-H.; Luh, T.-Y. J. Am. Chem. Soc. 2009, 131, 12579.
(b) Chen, C.-W.; Chang, H.-Y.; Lee, S.-L.; Hsu, I.-J.; Lee, J.-J.; Chen, C.-H.; Luh, T.-Y. Macromolecules 2010, 43, 8741.
(c) Yang, K.-W.; Xu, J.; Chen, C.-H.; Huang, H.-H.; Yu, J.-Y.; Lim, T.-S.; Chen, C.-H.; Luh, T.-Y. Macromolecules 2010, 43, 5188.
(d) Huang, H.-H.; Chao, C.-G.; Lee, S.-L.; Wu, H.-J.; Chen, C.-H.; Luh, T.-Y. Org. Biomol. Chem. 2012, 10, 5948.
(e) Yeh, N.-H.; Chen, C.-W.; Lee, S.-L.; Wu, H.-J.; Chen, C.-H.; Luh, T.-Y. Macromolecules 2012, 45, 2662. -
[48]
For a review, see: Luh, T.-Y. Acc. Chem. Res. 2013, 46, 378. doi: 10.1021/ar300170b
-
[49]
For reviews, see: (a) Cordova, A.; Rios, R. Angew. Chem., Int. Ed. 2009, 48, 8827.
(b) Schrock, R. R. Dalton Trans. 2011, 40, 7484.
(c) Gottumukkala, A. L.; Madduri, A. V. R.; Minnaard, A. J. ChemCatChem 2012, 4, 462.
(d) Schrock, R. R.; Hoveyda, A. H. Angew. Chem., Int. Ed. 2003, 42, 4592. -
[50]
(a) Jiang, A. J.; Zhao, Y.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2009, 131, 16630.
(b) Peryshkov, D. V.; Schrock, R. R.; Takase, M. K.; Müller, P.; Hoveyda, A. H. J. Am. Chem. Soc. 2011, 133, 20754.
(c) Marinescu, S. C.; Schrock, R. R.; Müller, P.; Takase, M. K.; Hoveyda, A. H. Organometallics 2011, 30, 1780. -
[51]
(a) Ibrahem, I.; Yu, M.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2009, 131, 3844.
(b) Banchet-Cadeddu, A.; Henon, E.; Dauchez, M.; Renault, J.-H.; Monneaux, F.; Haudrechy, A. Org. Biomol. Chem. 2011, 9, 3080. -
[52]
(a) Malcolmson, S. J.; Meek, S. J.; Sattely, E. S.; Schrock, R. R.; Hoveyda, A. H. Nature 2008, 456, 933.
(b) Sattely, E. S.; Meek, S. J.; Malcolmson, S. J.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2009, 131, 943.
(c) Yu, M.; Wang, C.; Kyle, A. F.; Jakubec, P.; Dixon, D. J.; Schrock, R. R.; Hoveyda, A. H. Nature 2011, 479, 88. -
[53]
(a) Flook, M. M.; Jiang, A. J.; Schrock, R. R.; Muller, P.; Hoveyda, A. H. J. Am. Chem. Soc. 2009, 131, 7962.
(b) Flook, M. M.; Gerber, L. C. H.; Debelouchina, G. T.; Schrock, R. R. Macromolecules 2010, 43, 7515.
(c) Flook, M. M.; Ng, V. W. L.; Schrock, R. R. J. Am. Chem. Soc. 2011, 133, 1784. -
[54]
Zhu, L.; Flook, M. M.; Lee, S.-L.; Chan, L.-W.; Huang, S.-L.; Chiu, C.-W.; Chen, C.-H.; Schrock, R. R.; Luh, T.-Y. Macromolecules 2012, 45, 8166. doi: 10.1021/ma301686f
-
[1]
-


图式 9 Schrock, Grubbs-Ⅰ, Grubbs-Ⅱ催化剂结构
Scheme 9 Structures of Schrock, Grubbs-Ⅰ, Grubbs-Ⅱ catalysts




图式 22 顺式间同排列聚降冰片烯形成机理示意图
Scheme 22 Mechanism for the formation of cis syndiotactic poly-norbornene
表 1 生物高分子与合成高分子比较
Table 1. Natural polymers vs. synthetic polymers
生物高分子a 合成高分子 分子量分布(PDI) 1 >1 单体重复性 无 有 单体序列性 有 无 手性 有 有(依赖手性辅基) 双股螺旋性 有 十分困难 复制性 有 未知 催化性 有 稀少 a蛋白质、核酸等.  下载: 导出CSV
下载: 导出CSV
-











































 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 24
- 文章访问数: 3376
- HTML全文浏览量: 338
 下载:
下载:
