
 Login In
Login In


Browse Articles
Call for papers
The Chinese Journal of Structural Chemistry, founded in 1982 by Prof. Jiaxi Lu, is an international peer-reviewed journal published in English. It publishes original research works about the structure and property of matter, including but not limited to coordination chemistry, organometallic chemistry, catalysis, energy, nanomaterial, theory/computation, structural characterization, pharmacy and life science. Published monthly by Fujian Institute of Research on the Structure of Matter, CAS, in the form of Articles, Communications, Reviews, Perspectives, and News & Views. The journal is published twelve issues a year by Fujian Institute of Research on the Structure of Matter, CAS and is available online:
https://www.sciencedirect.com/journal/chinese-journal-of-structural-chemistry
Contact information:
E-mail: cjsc@fjirsm.ac.cn; Tel: +86-591-63173769
Display Method: |
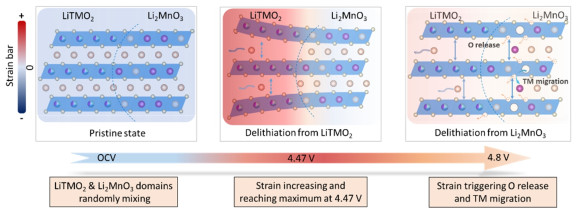
Self-powered X-ray detectors that combine high sensitivity with low detection limits have emerged as a critical focus in radiation detection research. This work pioneers the application of lead magnesium niobate-lead titanate (PMN-PT) single crystal, renowned for its piezoelectric properties, as a material in self-powered X-ray detectors. Due to its super internal electrostatic field, the PMN-PT single crystal self-powered detector exhibits a sensitivity of 346 μC Gyair-1 cm-2 and a detection limit of 0.43 nGyair s-1 under 40 keV X-rays, which is 17 times and 0.008% as those of the commercial α-Se detector, respectively. Notably, the detector maintains a high sensitivity of 157 μC Gyair-1 cm-2 even under hard 120 keV, surpassing the performance of most reported perovskite self-powered detectors. Furthermore, the detector exhibits a significantly reduced baseline drift (∼1.0 pA) and excellent operational stability in self-powered mode. This study conclusively demonstrates that polar piezoelectric crystals, which incorporate heavy metal elements, are highly promising candidates for developing high-performance self-powered X-ray detectors.
Antimony selenide (Sb2Se3) has emerged as a promising absorbing material in photocathode for photoelectrochemical (PEC) hydrogen evolution, exhibiting a development trajectory that outpaces many traditional semiconductors. Despite growing research interest, few reviews have provided a comprehensive roadmap for this class of photocathodes. In this review, starting from the scope and fundamentals of PEC systems, we will introduce how the quasi-one-dimensional (1D) crystal structure of Sb2Se3 enables anisotropic carrier transport, benign grain boundaries, and intrinsic stability. Advances in both vacuum and solution-based deposition techniques have yielded high-quality and scalable thin films, while recent progress in crystal orientation control, interface engineering, and co-catalyst integration has markedly improved efficiency. In parallel, the application of protection layers and bubble management strategies has extended operational stability. Beyond hydrogen evolution, Sb2Se3 has also shown potential for alternative solar-driven reactions. Nevertheless, challenges such as defect-induced recombination, interfacial mismatches, and long-term durability remain critical bottlenecks. Future progress will rely on integrated efforts in interface engineering, defect and crystal quality control, advanced protection and bubble management strategies, and the development of earth-abundant co-catalysts for alternative reactions, thereby linking fundamental material optimization with practical device deployment in solar-to-fuel technologies.
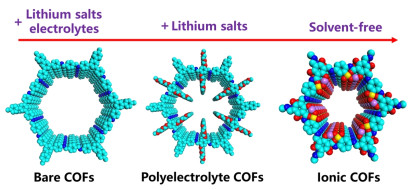
Nickel-cobalt (NiCo) alloys with bimetallic electronic modulation are considered promising candidates for urea electrolysis. However, their highly occupied d-band states limit the adsorption of reaction intermediates, resulting in insufficient activity toward urea oxidation reaction (UOR) and hydrogen evolution reaction (HER). Herein, a phosphorus-doped amorphous NiCo alloy is synthesized, in which P incorporation modulates the electronic structure and upshifts the d-band center of NiCo alloy, thus optimizing the adsorption of OH− and urea molecules. The sample with optimal P content exhibits low potentials for UOR (E10/1000 = 1.28/1.65 VRHE) and HER (E−10/−1000 = −67/−206 mVRHE). Furthermore, it achieves an industrial-level current density of 500 mA cm−2 at a cell voltage of 1.72 V in the membrane electrode assembly, while maintaining stable activity for 150 h. Therefore, this work provides a new strategy to optimize reactant adsorption and ultimately enhance the electrocatalytic activity by modulating the d-band center.
The construction of bifunctional photocatalysts that integrate oxidative and reductive sites for concurrent CO2-to-CH4 and H2O-to-H2O2 conversion under visible light remains a formidable challenge. Herein, a facile photo-deposition route was designed to support Pd nanoparticles (4.5 nm) onto SnNb2O6 nanosheets (3.5 nm) for constructing a photocatalyst (Pd-PNS). Under visible-light irradiation (λ ≥ 400 nm) and without any sacrificial agent, the optimized Pd-PNS catalyst achieves CH4 and H2O2 evolution rates of 24.3 and 79.2 μmol g-1 h-1, respectively (1:4 stoichiometry), with an apparent quantum efficiency (CH4) of 0.40 % at 420 nm, outperforming most reported systems operated under equivalent conditions. The results of characterizations reveal the formation of a strong metal-support interaction (SMSI) and a Schottky junction between Pd and SnNb2O6. SMSI causes the loss of lattice oxygens, thus generating Nb4+ and O vacancies. The Schottky junction induces the migration of photogenerated electrons to Pd NPs, while holes are trapped at lattice oxygen sites (around Nb4+), thereby improving charge transfer and separation. CO2 is reduced to CH4 on Pd NPs, while H2O is oxidized to H2O2 on Nb4+ sites, validating a true dual-site photocatalytic cycle (CO2 + 6 H2O → CH4 + 4H2O2). This work offers a valuable strategy for sacrificial-free co-production of solar fuel and green oxidant by photoinducing SMSI in a photocatalyst.
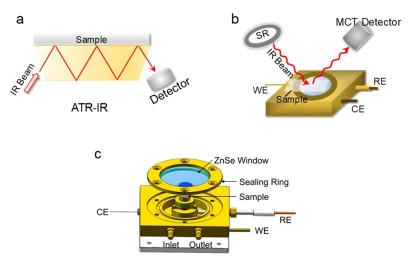
As food safety concerns grow, controlling organophosphorus pesticide residues and food spoilage substances has become a priority. Effectively degrading contaminants like dichlorvos (DDVP) on chili peppers and histamine (HA) in sardines is crucial for food safety. This study explores the rapid degradation and mechanism of DDVP on chili pepper surfaces and HA in sardines using S-scheme Al6Si2O13/g-C3N4 (ASO/CN) nanocomposites. Rapid detection methods analyzed standard solutions of DDVP and HA. The synthesized ASO/CN nanocomposites showed excellent photocatalytic activity, reducing DDVP from 100% to 17.7% in 100 min, outperforming individual ASO and CN. The catalyst maintained high degradation efficiency over five cycles (500 min). The ASO/CN nanocomposites also degraded HA in sardines, lowering it from 100% to 9.2% in 70 min, with stable performance over five cycles (350 min). Characterization techniques, including in situ X-ray photoelectron spectroscopy (XPS), Kelvin probe force microscopy (KPFM), femtosecond transient absorption spectroscopy (fs-TAS), and differential charge density calculations, confirmed an S-scheme photocatalytic mechanism that enhanced radical formation. Based on these findings, practical tests under natural conditions were conducted. The catalyst reduced DDVP on chili peppers from 100% to 39.2% over seven days, outperforming the control (100% to 78%). In sardines, HA in the ASO/CN-treated group increased from 25.1% to 71.6% over five days, while the untreated group increased from 25% to 90.5%. These results offer a new approach for organophosphorus pesticide degradation and meat preservation.
Mn-activated phosphors have attracted great attention, but the inevitable self-reduction of Mn4+ to Mn2+ and precise regulation of Mn4+/Mn2+ content remain serious challenges. Herein, through solid-state reaction in air, we demonstrate that the degree of self-reduction and valence of Mn can be accurately manipulated in normal spinel ZnGa2O4 (ZGO) by incorporation of Li+ and F-, achieving color-tunable photoluminescence (PL) as desired, from blue self-luminescence to red/green emission of Mn4+/Mn2+. Both theoretical (i.e., density functional theory, bond energy theory) and experimental (i.e., dynamic/static spectroscopy) analyses indicate that Li+ occupying the tetrahedral Zn2+ site can push Mn4+ into the octahedral Ga3+ site and restrain self-reduction of Mn4+ owing to localized charge accumulation around Li+ that depletes excess electrons. Furthermore, F− substitution can repair intrinsic oxygen vacancies, further suppressing self-reduction and detrimental electron-capturing effects. Meanwhile, Li+/F− incorporation can distort ZGO and break the forbidden transition of Mn4+, leading to broadened PL and enhanced efficiency. The long-persistent PL (LPL) of ZGO: Mn/Li/F with wide shallow/deep traps is also explored in depth. Finally, the Li/F-dependent tunable PL and LPL of ZGO: Mn/Li/F show great potential for applications in ratiometric optical thermometers, plant lighting, white LEDs, and dynamic anticounterfeiting.
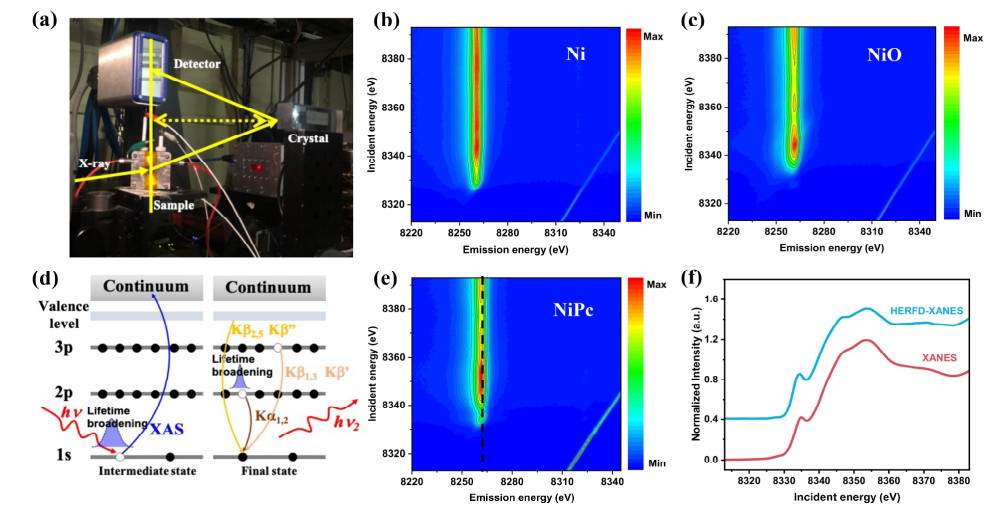
Photocatalytic synthesis of hydrogen peroxide (H2O2) from water and oxygen is a promising yet challenging green route, primarily limited by severe charge recombination and the inefficient activation of inert O2 molecules. To address these dual bottlenecks, this work constructs an organic/inorganic step-scheme (S-scheme) heterojunction by intimately coupling C3N4 with Bi2O3. This unique architecture, as deciphered by in-situ XPS and femtosecond transient absorption spectroscopy, drives efficient S-scheme charge transfer. This process not only achieves spatial separation of powerful photogenerated carriers but also retains their high redox potential. Crucially, density functional theory calculations reveal that the interfacial electronic coupling induces a significant electron redistribution, which dramatically enhances the adsorption and activation of O2 molecules—a finding corroborated by oxygen temperature-programmed desorption. Consequently, the optimized C3N4/Bi2O3 photocatalyst delivers a remarkably high H2O2 production rate of 4.03 g-1 h-1 under simulated sunlight. The in-situ generated H2O2 further translates into superior disinfection efficacy, achieving 99.9% inactivation of E. coli within 60 min. This work elucidates the charge dynamics at organic/inorganic S-scheme interfaces and showcases a viable pathway for designing efficient photocatalysts for coupled solar fuel production and environmental applications.
Efficient spatial separation and orderly migration of charge carriers, as well as robust kinetics at catalytic sites, constitute the fundamental and core issues in enhancing the conversion of solar energy into hydrogen from water splitting. Herein, surface localization polarization engineering has been confirmed to be effective to accelerate the oriented charge migration dynamics behaviour and collaboratively activate redox crystal facets on the constructed Co3O4/In2S3 S-scheme heterojunction. Theoretical calculations and ultrafast atomic-scale spatiotemporal analysis demonstrate that, Co3O4/In2S3 heterojunction endows the surface localized field pointing from the hexagonal In2S3{102} to cubic Co3O4{111} with an increased field strength on the basis of Co3O4 from 0.27 μV to 2.19 μV and lifetime from 260.99 ns to 975.18 ns for charge carrier transfer to the surface of the redox crystal facets. Polarization-state charge uneven distribution reduces and enhances the binding energy of components on active crystal facets thus with correspondingly increasing and decreasing onsite electron density for promoting chemical adsorption of *H and *OH, respectively. Solar to H2 of 1.32% at AM 1.5G is achieved along with high photocatalytic stability. Surface polarization engineering plays a pivotal role in our study, enabling substantial tuning of charge transfer behavior and crystal facet surface activation within S-scheme heterojunctions for the improved photocatalytic H2 generation.
Hydrogenolytic debenzylation is a crucial step to restore the activity of the protected functional groups for targeted compounds while avoiding the formation of by-products. However, conventional methods suffer from low catalytic efficiency due to slow mass transport and/or severe leaching or agglomeration of active species. It highlighted the urgent need to develop catalysts that enable the coexistence of high mass transport and abundant active sites. In this work, as a proof of concept, a series of bimetallic PdM catalysts (M=Fe, Ni, Cu) anchored on hierarchically ordered porous skeletons were developed to enable the fast hydrogenolytic debenzylation of tetraacetyldibenzylhexaazaisowurtzitane (TADBIW) into tetraacetylhexaazaisowurtzitane (TAIW). We demonstrate that a hierarchically ordered macroporous N-doped carbon (DOM-NC) scaffold can simultaneously guarantee rapid diffusion of the bulky substrate (TADBIW) and stabilize electron-rich Pd(0) species when a trace amount of Ni is co-introduced. The resulting PdNi/DOM-NC catalyst delivers 97.3 % yield of TAIW within 2.5 h at an ultra-low Pd loading (Pd: TADBIW = 2.5 wt ‰) and retains >95 % activity after four cycles without detectable Pd loss. Structural and theoretical analyses reveal that (i) the 3DOM architecture shortens diffusion lengths, (ii) adjacent Ni atoms transfer electron density to Pd, increasing the surface Pd(0) fraction, and (iii) the Ni–Pd synergy lowers the H2 dissociation barrier and accelerates the rate-determining second debenzylation step. This study provides new insights into the rational design of highly efficient noble-metal-based heterogeneous catalysts and offers guidance for developing catalysts for other macromolecular hydrogenolysis reactions.
The incorporation of a third component to ternary organic solar cells (T-OSCs) is an effective strategy for enhancing the power conversion efficiency (PCE). While the fluorination extent of acceptor molecules is critical for modulating energy levels, molecular packing, and blend compatibility, systematic studies on the fluorination pattern remain scarce. In this work, we synthesized three novel fluorinated acceptors (o-2F, p-2F, and t-4F) with varied fluorine substitution patterns on the central benzene core via an atom-economical direct C–H arylation route. The influence of the fluorine substitution motif on molecular conformation, aggregation behavior, and device performance was systematically investigated. Theoretical calculations revealed that increased fluorination significantly enhances molecular planarity. Device performance tests demonstrated that the ortho-difluorinated acceptor o-2F promotes favorable nanoscale phase separation and facilitates efficient charge transport. Consequently, the PM6:Y6:o-2F-based TOSCs achieved a notable improvement in PCE from 16.16% to 17.41% compared to the PM6:Y6 binary counterpart, accompanied by concurrent enhancements in open-circuit voltage (from 0.83 V to 0.85 V), short-circuit current (from 26.45 to 27.19 mA cm-2), and fill factor (from 73.56% to 75.33%). In contrast, the excessively fluorinated t-4F led to overly enhanced crystallinity, resulting in limited morphological optimization and marginal PCE improvement. This study underscores the importance of balancing molecular planarity and aggregation through rational fluorination design, providing valuable guidance for developing high-performance acceptor materials for T-OSCs.


