 图1
核酸适配体与药物的结合方式[59]
Figure1.
Schematic illustration of combination between aptamer and drug[59]
图1
核酸适配体与药物的结合方式[59]
Figure1.
Schematic illustration of combination between aptamer and drug[59]
引用本文:
堵玉林, 梁静. 核酸适配体在肿瘤靶向治疗方面的研究进展[J]. 化学通报,
2017, 80(9): 809-818, 862.

Citation: Du Yulin, Liang Jing. Advances in the Application of Aptamers in Tumor Targeted Therapy[J]. Chemistry, 2017, 80(9): 809-818, 862.
Citation: Du Yulin, Liang Jing. Advances in the Application of Aptamers in Tumor Targeted Therapy[J]. Chemistry, 2017, 80(9): 809-818, 862.
核酸适配体在肿瘤靶向治疗方面的研究进展
摘要:
传统的抗肿瘤药物大多不具有选择性,在临床治疗中产生了严重的毒副作用。核酸适配体是一种小分子核酸,能够与靶标高亲和性、高特异性地结合。选择与癌症发生发展过程密切相关的生物标记物为靶标进行SELEX过程筛选出的核酸适配体自身可作为药物,也可与药物、siRNA、纳米粒等结合构成靶向给药体系,该体系能靶向作用于特定的肿瘤细胞,降低对正常细胞的毒性,用药量显著降低,药效提高。本文综述了近年来核酸适配体直接作为抗肿瘤药物、药物载体、siRNA载体以及作为纳米材料靶向剂构成多元复合靶向给药体系在肿瘤靶向治疗领域的研究进展。
English
Advances in the Application of Aptamers in Tumor Targeted Therapy
Abstract:
Traditional anti-tumor drugs can cause serious side effects in clinical treatment due to their nonspecific toxicity. Aptamers are a class of small nucleic acid ligands that have high affinity and specificity for their targets. Well-characterized biomarkers, especially those closely related to the development of cancer, can be used as targets for aptamer selection via the SELEX method. The newly obtained aptamers may be able to act as drugs themselves, or conjugate with other chemical drugs, siRNA, and nanoparticles to generate targeted drug delivery systems which can target specific tumor cells, thus minimizing the toxicity to normal cells, reducing the dose needed for treatment and enhancing therapeutic efficacy. In this review, we summarize the current advances in the application of aptamers in tumor targeted therapy with aptamers as anti-tumor drugs, as drug carriers, as RNA-based therapeutic carriers or as targeted ligands for conjugates with nanoparticles.
-
Key words:
- Aptamer
- / Anti-tumour drug
- / Targeted therapy
- / Nanomaterial
- / siRNA
-
核酸适配体(Aptamer)的名称来源于拉丁词“aptus”,意为“适合”,最早由Ellington等[1]和Tuerk等[2]在1990年提出,是通过指数富集配体系统进化技术(Systematic evolution of ligands by exponential enrichment,SELEX)从化学合成的1012~1015个随机单链寡核苷酸(ssDNA或RNA)文库中筛选得到。当靶标存在时,核酸适配体可经过自身卷曲、折叠形成特定的三维构型,如发夹、凸环、四角环等,通过范德华力、氢键、静电作用、碱基堆积力等与靶标高亲和性、高特异性地结合,这一过程类似抗体-抗原的结合,因此核酸适配体又被称为“化学抗体”。
尽管与抗体功能相似,核酸适配体仍具有自身独特的优势,如分子量低,易于穿透组织;无免疫原性,不引起机体的免疫反应;热力学稳定性好,可反复变性、复性,不引起三维构型的改变;易于合成,易于修饰,合成过程可自动化[3, 4];可以使用互补的寡核苷酸链作为解毒剂终止核酸适配体的作用[5, 6]。但核酸适配体也存在一些天然的劣势,如易于被血液中的核酸酶降解[7],因分子量低易于被肾脏排泄清除出体外,造成体内作用时间较短。除少量的核酸适配体具有天然的抗核酸酶水解能力外[8],大多数由SELEX技术得到的核酸适配体必须经过一定的化学修饰,才能用于临床,如在3’端和5’端进行修饰[9]可抵抗核酸外切酶的降解;核糖或脱氧核糖的2’位H用-NH2[10, 11]、-OCH3[12, 13]、-F[14, 15]等取代,或对其进行磷硫酰化,将磷酸二酯键修饰为硫代磷酸二酯键可抵抗核酸内切酶的进攻;在5’位连入20~40kDa聚乙二醇[16, 17]或胆固醇[18]等提高分子量,延长血液循环时间。除此之外,锁核酸(Locked nucleic acid)技术[19]和镜像(Spiegelmers)技术[20, 21]等也可增加核酸适配体的稳定性。
化学修饰可以在SELEX过程前、过程中和过程后进行,这些化学修饰虽能提高核酸适配体的稳定性,但可能会影响其与靶标的结合能力,因此修饰后的核酸适配体还需再次检测与靶标结合的亲和性和特异性。
核酸适配体的靶标形式多样,包括各种小分子、蛋白质、细胞[22, 23]、组织[24]甚至是有机体[25, 26]。20世纪90年代,以一些具有重要临床价值的肿瘤标记物或生物标记物为靶标,如温韦伯氏因子(von Willebrand Factor,vWF)[27, 28]、血小板源生长因子(Platelet-derived growth factor,PDGF)[29, 30]、E-选择素(E-selectin)[31, 32]、血管内皮生长因子(Vascular endothelial growth factor,VEGF)[33, 34]、核因子kB(Nuclear factor kB,NFkB)[35, 36]、细胞粘合素C(Tenascin-C)[37, 38]和前列腺特异性膜抗原(Prostate specific membrane antigen,PSMA)[39, 40]等,筛选出的核酸适配体在肿瘤的靶向研究方面获得了迅速发展,有望用于肿瘤诊断[41~43]、肿瘤成像[44, 45]、肿瘤治疗[46, 47]等,下面仅就核酸适配体在肿瘤靶向治疗方面的应用作简单概述。
1 核酸适配体作为靶向药物
1.1 Pegaptanib
2004年11月,由Eyetech与辉瑞公司共同研发生产的Pegaptanib(商品名Macugen)[48]成为第一个获得美国食品和药物管理局(FDA)批准的抗血管内皮生长因子的核酸适配体药物,用于治疗老年性黄斑变性。血管内皮生长因子VEGF是一种在生理性和病理性血管新生时起重要作用的蛋白质,它与相应受体(VEGF Receptor,VEGFR)结合促进下游信号通路转导,进而促进新生血管形成,同时增加血管通透性。
通过可变剪接,VEGF基因主要表达4种VEGF亚型,分别具有121(VEGF121)、165(VEGF165)、189(VEGF189)、206(VEGF206)个氨基酸,其中VEGF165[49]是造成老年性黄斑变性和糖尿病黄斑水肿中非正常性血管新生的重要因素。Macugen仅靶向作用于VEGF165,经玻璃体腔注射后,Macugen与VEGF165的肝素结合区相互作用,阻碍了VEGF165与其受体结合,避免受体被激活引起下游反应,从而抑制新生血管的生成[50]。Eyetech制药公司[51]通过伊文思蓝检测体外研究大鼠角膜新生血管模型和小鼠早产儿视网膜病变模型,证实Macugen具有抑制VEGF的作用,从而降低血管通透性、抑制眼部病理性新生血管形成。随后发表的Macugen IA期临床报告显示,注射Macugen 3个月后,80%的患者表现稳定或视力好转,且安全性高。然而,Macugen并不能完全抑制眼部病理性新生血管的生成,对视力提高的程度有限,主要原因在于Macugen仅能与VEGF165结合,不能结合并抑制其他活性VEGF亚型。
基于Macugen抑制新生血管生成这一特性,Huang等[52]利用肾内接种的Wilms瘤模型,研究了Macugen的抗肿瘤疗效,发现Macugen还可以有效抑制Wilms肿瘤的生长和转移。
1.2 AS1411
AS1411是富含鸟嘌呤的DNA核酸适配体,含有26个核苷酸片段,被认为是第一个进入临床研究的抗肿瘤核酸适配体。AS1411是基于细胞筛选得到的,它的作用机制并不十分清楚,通常认为它高选择性、高特异性地与位于肿瘤细胞表面的核仁蛋白结合,而后被细胞内吞,与细胞质内的核仁蛋白结合,抑制核因子kB[53],使抗细胞凋亡的BCL-2蛋白(B-cell Lymphoma protein)的mRNA去稳[54],从而使肿瘤细胞死亡。Reyesreyes等[55]研究了AS1411的作用机制,他们认为AS1411与核仁蛋白结合,持续激活Rac1蛋白,过度刺激巨胞饮,引起细胞死亡。
AS1411用于多种临床前研究和动物模型中,被证实对人类乳腺癌、肺癌、胰腺癌和急性骨髓性白血病均有效[56, 57]。目前AS1411正处于急性骨髓性白血病的Ⅱ期临床研究。
1.3 NOX-A12
NOX-A12[46]由镜像技术[20, 21]发展而来。天然核酸由D-核糖或D-脱氧核糖和碱基、磷酸构成的核苷酸组成,由L-核糖或L-脱氧核糖构成的核酸则不被核酸酶识别,抗核酸酶水解的稳定性提高。NOXXON药物公司发展了镜像技术,首先以真正靶标的镜像分子为SELEX过程的靶目标,在D-核糖或D-脱氧核糖构成的寡核苷酸文库中筛选,得到对镜像靶标具有亲和性和特异性的核酸适配体,对其进行测序,然后重新合成碱基序列完全相同但糖基为L-核糖或L-脱氧核糖的L-核酸适配体,它不但可与真正的靶标高亲和性、高特异性地结合,而且37℃下可在血液中稳定存在60h以上不被核酸酶降解,增加了血液循环时间。
趋化因子CXCL12(Chemokine C-X-C motif ligand 12) 在促进肿瘤增殖转移、促进血管新生、延缓肿瘤凋亡等过程中起着关键作用,它有两个重要区域,一是与趋化因子受体CXCR4、CXCR7的结合区域,另一处是在细胞上的非特异性着落点。NOX-A12靶向作用于CXCL12,它与CXCL12的两个区域均能结合,从而抑制CXCL12和肿瘤细胞的相互作用。Fearon等[58]认为NOX-A12的抗癌机理在于NOX-A12能调节肿瘤细胞的微环境,例如CXCL12能吸引血癌细胞到骨髓缝隙中隐藏,而NOX-A12则驱逐这些肿瘤细胞进入外周血液,抗肿瘤药物可以有效抵达;CXCL12在一些实体瘤周围形成具有保护作用的生物屏障,阻止免疫细胞进入,NOX-A12则能破坏这道屏障,使免疫细胞进入肿瘤组织中,杀死肿瘤细胞;CXCL12可以吸引一些修复细胞到达被放化疗损伤的肿瘤组织中,NOX-A12阻止这种作用,使肿瘤不能被修复,进而死亡。
已证实NOX-A12对结肠癌、胰腺癌、脑癌、多发性骨髓瘤均有效,NOX-A12在美国被授予恶性胶质瘤的孤儿药资格认定,在欧洲被授予神经胶质瘤的孤儿药资格认定。
2 基于核酸适配体-药物分子的靶向给药体系
传统的肿瘤治疗方法包括化学治疗、放射治疗、光热治疗等,因不具有靶向作用,不仅杀死癌细胞,同时也损害正常细胞,引起胃肠道反应、骨髓抑制、免疫功能低下等严重的副作用。因此,将核酸适配体与抗肿瘤药物结合,不仅可将药物特异性靶向病变部位,提高药物疗效,降低用药量,还能减少对正常细胞的损伤。核酸适配体与药物结合的方式有两种:非共价结合(嵌入)和共价结合。
2.1 非共价结合(嵌入)
核酸适配体和药物无须经过任何化学修饰,可通过氢键、静电力、范德华力等非共价相互作用结合,形成简单、有效的靶向给药体系。
阿霉素(Dox)是一种常见的抗肿瘤药物,能有效治疗白血病、淋巴癌、肝癌等多种恶性肿瘤。因具有平面四环结构,可嵌入肿瘤细胞的DNA碱基片段(特别是CG/GC),阻碍DNA的复制,达到抗肿瘤目的。Dox的主要缺点是具有强烈的细胞毒性,严重损害正常细胞。根据Dox能嵌入核酸结构这一特性,可将Dox嵌入富含CG/GC序列的核酸适配体中,形成Apt-Dox靶向给药体系,降低Dox对正常细胞的毒性。
核酸适配体A10是由71个核苷酸组成的单链RNA,能特异性靶向人前列腺癌细胞LNCaP上过表达的PSMA蛋白。Bagalkot等[60]通过非共价相互作用将Dox嵌入A10的三维结构得到A10-Dox复合体,能够将Dox靶向输运到PSMA过表达的人前列腺癌细胞LNCaP。由于A10-Dox药物是通过非共价相互作用连接,A10和Dox均可保持高的生物活性,不会降低药效。
Hu等[61]筛选出一种含86个碱基的靶向mucin 1(MUC1) 的核酸适配体MA3,能够识别MUC1阳性的肿瘤细胞MCF7。他们将Dox嵌入MA3的DNA结构形成MA3-Dox复合体,发现该复合体能以受体介导的内吞作用进入细胞,对MUC1阳性的肿瘤细胞具有靶向杀伤作用。
人类表皮生长因子受体2(Human epidermal growth factor receptor 2,HER2) 是一种膜受体,它的过表达往往与肿瘤的发生发展密切相关。核酸适配体HB5能够选择性结合HER2阳性的乳腺癌细胞,Liu等[62]通过将Dox嵌入核酸适配体HB5,构建了HB5-Dox靶向给药体系,将Dox靶向输运到HER2阳性的乳腺癌细胞,降低了Dox对HER2阴性细胞的杀伤作用。
2.2 共价结合
虽然通过非共价结合形成ApDC(Aptamer-drug conjugation)简单有效,但是许多药物并不能有效嵌入核酸适配体中,而且药物的嵌入可能会改变核酸适配体的结构,影响核酸适配体对靶标的识别和特异性结合,这时需要将核酸适配体与药物以共价键直接相连或通过功能性连接体相连。共价结合是核酸适配体与药物形成ApDC最常见的形式,形成的ApDC较非共价结合稳定,在到达靶标前药物与核酸适配体不会发生解离,提高了靶向给药效率。此外,对连接体进行精心设计,还可以增加药物的负载量。
DNA核酸适配体sgc8c能特异性靶向人急性淋巴细胞白血病T淋巴细胞(CCRF-CEM)表面上过表达的酪氨酸蛋白激酶7(Protein tyrosine kinase 7,PTK7)。Huang等[63]将Dox与sgc8c通过腙键共价相连形成sgc8c-Dox复合物,该复合物能特异性靶向CCRF-CEM细胞,并以受体介导的内吞作用进入肿瘤细胞内涵体中。由于腙键具有pH敏感性(对酸不稳定),在内涵体的酸性环境中易断裂释放出药物Dox,抑制肿瘤细胞的生长,且药效与单独使用Dox时相同。
为进一步提高药物负载率,Zhu等[64]设计了aptNTrs(aptamer-tethered DNA Nano-Trains)靶向给药体系。对DNA核酸适配体sgc8的5’端进行修饰,引入DNA触发探针(DNA trigger probe)。修饰后的sgc8-trigger作为“火车头”靶向CCRF-CEM细胞表面的PTK7。两个DNA单体(M1和M2) 在不接触触发探针时,分别以独立的发夹型结构存在。一旦在M1和M2混合体系中引入sgc8-trigger时,则立即引起M1和M2构型改变,发夹型结构解体,M1与触发探针、M2与M1间的互补碱基相互杂交、聚合,构成“车厢”运载药物。自组装完成后,aptNTrs体系不但显示高的载药量,药物:sgc8c-NTrs为50:1,而且仍然可被CCRF-CEM细胞内化,表现出较强的抑制肿瘤细胞增殖的能力。
除此之外,Tan等[65]将光敏剂Ce6交联到核酸适配体TD05上,TD05能特异性识别Burkitt淋巴瘤细胞,从而使光化学治疗具有靶向性。
3 基于核酸适配体-siRNA的靶向药物
小分子干扰RNA(Small interfering RNA,siRNA)通常是一段长约21个核苷酸的双股RNA(dsRNA),由长的双链RNA或小发夹RNA(Small hairpin RNA, shRNA)在Dicer酶作用下加工而成。在细胞质内,siRNA双股链中的一条链与包括argonaute蛋白在内的相关蛋白质因子结合形成RNA诱导的沉默复合体(RNA-induced silencing complex, RISC),通过碱基配对识别与siRNA互补的靶标mRNA,在Mg2+与ATP的参与下,argonaute蛋白利用其核酸内切酶的活性切割靶标mRNA,产生5’磷酸端和3’羟基端片段,使之更易遭受核酸外切酶的攻击进一步降解。
siRNA只沉默与其序列互补的mRNA,能特异性抑制致病基因的表达,从源头上阻止肿瘤的产生和发展。要发挥RNA干扰功能,siRNA首先需要进入靶细胞的细胞质,但siRNA易被血液中的核酸酶降解,对特定组织或细胞缺乏靶向性,且不易透过细胞膜,自身较难进入细胞质中,这成为发展基于RNA疗法的制约因素之一[66]。除了常见的载体如脂质体、聚合物、糖类、蛋白质等,核酸适配体为siRNA的靶向输运提供了一种独特高效的工具。
McNamara等[67]发展了一种aptamer-siRNA复合体,将Plk1 siRNA或Bcl2 siRNA与修饰后的RNA核酸适配体A10共价结合,发现这种复合物的形成并未影响A10和siRNA的生物功能,A10能特异性结合到PSMA过表达的CEM细胞,而两个siRNA则分别使Plk1基因和Bcl2基因沉默,显著抑制了肿瘤的生长。
HIV是严重危害公众健康的恶性疾病,感染HIV-1病毒后,病毒侵入宿主,它的外壳蛋白gp120识别辅助性T细胞表面的CD4受体,与之结合并引发膜融合,随后病毒将RNA和酶等注入辅助性T细胞,被感染的细胞也会在细胞表面表达gp120,引发更多的辅助性T细胞感染。Zhou等[68]将靶向HIV gp120的RNA核酸适配体进行2’-F修饰,与作用于HIV-1tat/rev区域的siRNA连接形成复合物,该复合物对HIV-1具有双重抑制功能,不仅能特异性结合gp120,进入表达gp120的细胞,而且使相应的tat/rev靶基因沉默。该复合物能强效且持续地抑制T细胞内HIV-1病毒的复制,不引发干扰素反应,推进了HIV核酸类药物的研究。
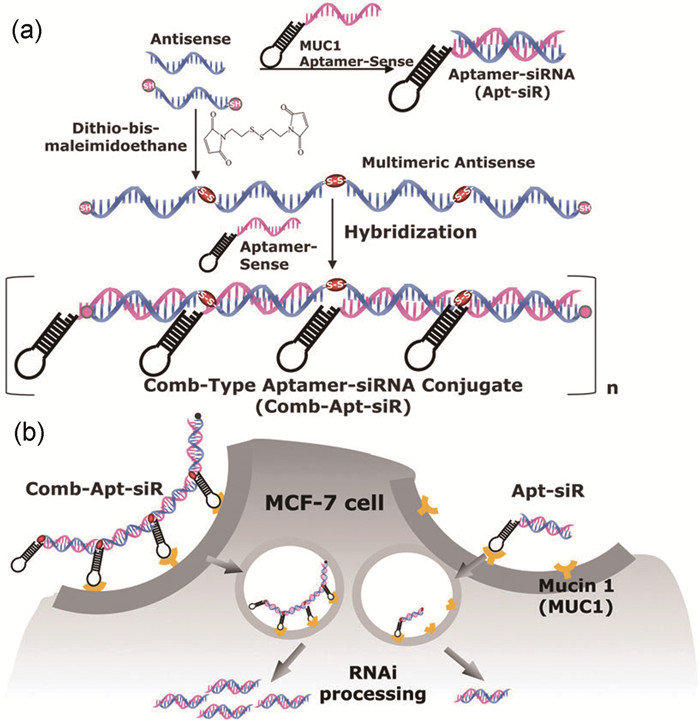
MUC1通常在恶性腺癌(adenocarcinomas)细胞表面过表达,Yoo等[69]将靶向MUC1的DNA核酸适配体连接siRNA正义链,将两端均有巯基修饰的单条siRNA反义链采用化学交联剂DTME连接成线性siRNA多聚物,通过siRNA的正义链与反义链间的碱基互补配对,首次设计出一种多价、具有梳状结构的Comb-Apt-siR复合物(图 2)。该复合物仅包含功能性siRNA和DNA核酸适配体,大大降低了由外源性核酸引起的非特异性免疫。研究结果表明,该复合物不仅特异性靶向MUC1过表达的癌细胞,如MCF-7、A549等细胞,而且梳状多齿结构能同时和细胞膜上的MUC1蛋白结合,增加了Comb-Apt-siR复合物与靶细胞间的亲和力,提高了靶细胞对梳状复合物的吸收(通过细胞内吞),有效输运siRNA到细胞质中并沉默靶标mRNA,抑制肿瘤细胞增殖,且不会对机体产生任何毒性。
Tang等[70]对核酸适配体介导的脂质体靶向输运siRNA进行了研究,将靶向PSMA的核酸适配体A10-3.2与脂质体偶联,用于靶向输运Bcl2 siRNA到PSMA过表达的LNCaP细胞。结果表明,与单一脂质体输运siRNA相比,核酸适配体介导的输运体系对PSMA过表达的LNCaP细胞表现出更高的输运效率,对Bcl2基因的抑制作用也更加显著,有效抑制了前列腺癌的发展。
RNA疗法尚在早期阶段,脱靶、免疫反应、siRNA或miRNA的有效输运等因素均制约了它的临床前和临床研究。而核酸适配体具有靶向性、自身无免疫原性,通过受体介导的内吞可进入细胞质中,是较为理想的siRNA载体,基于Apt-siRNA复合体的研究将会进一步促进RNA疗法的发展。
4 基于核酸适配体-纳米材料的靶向药物
因核酸适配体分子量小,在血液循环中的半衰期短,严重影响了药效。将它与纳米颗粒结合,不但能有效延长在血液循环中的半衰期,而且纳米颗粒大的比表面积还能提高药物和核酸适配体的负载量,均匀一致的形貌尺寸也使其呈现出良好的生物分布,因此核酸适配体-纳米颗粒是一种具有应用前景的靶向给药体系。
4.1 核酸适配体-金纳米颗粒
金纳米颗粒(AuNPs)具有特殊的光电性能、高的稳定性以及良好的生物相容性,其粒径小,形态可控,能通过渗透和滞留增强效应(Enhanced permeability and retention effect,EPR)优先在肿瘤部位富集。金纳米颗粒表面易于功能化,引入巯基修饰后可实现药物控释,因此许多研究者将金纳米颗粒与核酸适配体结合,用于药物的主动靶向输运。
由于金-银纳米颗粒特殊的光热效应,Huang等[71]将其与特异性识别CCRF-CEM细胞的核酸适配体sgc8c偶联构成Apt-NPs复合物,可用于光热治疗。实验结果表明,Apt-NPs复合物不仅能靶向肿瘤细胞,在近红外光照射下AuNPs温度不断升高从而杀死肿瘤细胞,但对正常细胞基本不造成损伤(存活率达87%),且大大降低了所需的激光照射量。
Luo等[72]设计了一种新型靶向给药体系,可通过激光照射控制药物的精准释放。他们用发夹型DNA(hpDNA)装载Dox,将其与核酸适配体sgc8c共同连接到AuNPs表面形成Apt/hp-Dox-AuNPs复合物。当sgc8c特异性靶向并进入CCRF-CEM细胞后,该体系在特定波长(532nm)的激光作用下会释放出Dox,对肿瘤细胞起到杀伤作用。
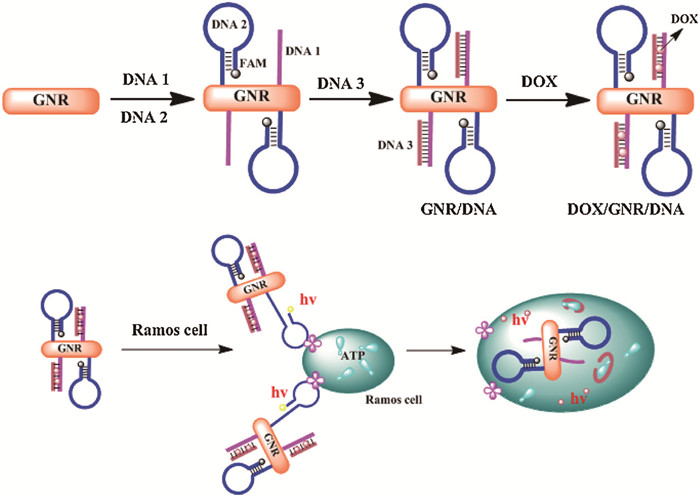
Guo等[73]构建了一种基于金纳米棒的双重核酸适体探针平台,用于肿瘤的靶向治疗。为实现这一平台对靶细胞的特异性识别,他们将一端带有FAM荧光团修饰的Romas-核酸适配体(DNA2) 连接到金纳米棒(GNRs)表面,赋予其靶向Romas细胞的能力。当核酸适体构象改变时,荧光信号会随之被猝灭或恢复,因而可用作刺激响应性细胞传感器。与此同时,将富含GC序列的三磷酸腺苷(ATP)-核酸适配体(DNA1) 也修饰在GNRs表面,由于ATP-核酸适配体为DNA双链结构,可有效提高DOX的装载量。这一平台不仅能特异性靶向Romas细胞并进入靶细胞内部,还能有效检测细胞内的ATP分子,当核酸适配体与ATP特异性结合后释放出药物分子Dox,进一步控制药物的精准释放,抑制肿瘤的生长。
4.2 核酸适配体-介孔SiO2纳米颗粒
介孔SiO2纳米颗粒(MSNs)具有良好的生物相容性、高的热稳定性、表面易于修饰、孔径规则且在2~50 nm范围内可连续调节等特性,被广泛用作药物载体。因其拥有巨大的比表面积和比孔容,能有效提高药物的负载量,延长药物的作用时间。将核酸适配体修饰到MSNs表面可实现药物的靶向输运及定点释放。Yang等[74]用介孔SiO2包裹GNRs,并在MSNs表面引入3’端延伸了12个碱基(DNA-2) 的核酸适配体AS1411作为封堵剂,经此修饰的MSNs依次与Dox、DNA-1孵育,DNA-1也是一条含12个碱基的寡核苷酸链,它与DNA-2互补配对可将药物Dox封堵于介孔SiO2的孔道中。在近红外光照射下,由于GNRs的光热效应,MSNs内温度不断升高,导致DNA-1与DNA-2互补碱基对间的氢键断裂,孔道开放,释放出Dox。体外实验中将此MSNs-Dox体系分别与乳腺癌细胞MCF-7以及正常细胞NIH3T3孵育,发现核酸适配体AS1411可有效靶向乳腺癌细胞MCF-7,在近红外光照下释放Dox,有效杀死MCF-7细胞,实现药物的靶向输运和光控释放。其中,核酸适配体AS1411既是靶向剂,又是封堵剂,充当对温度敏感的“纳米阀”,在体温下存留药物,在高温下释放药物,实现“温控释药”。而温度的改变则是依赖于GNRs对近红外光的光热效应,将光能转变为热能,在实际操作上实现了“光控释药”。
Zhao等[75]将ATP-aptamer与两段5’端炔基修饰的短单链DNA(臂ssDNA1和臂ssDNA2) 互补杂交形成三明治结构,通过环加成反应将臂ssDNA1和臂ssDNA2共价连接到叠氮基功能化的MSNs表面,与之互补的ATP-aptamer也随之被固定到MSNs表面,构建了一种新型的三明治型DNA封堵介孔SiO2的ATP响应控释体系。他们又在ATP-aptamer两端各延长7个碱基,通过延长的碱基与短单链DNA(ssDNA)进行碱基互补配对形成三螺旋结构,将这种三螺旋DNA作为封堵剂、MSNs作为载体,首次构建出一种三螺旋DNA封堵介孔SiO2的ATP响应控释体系。这两种体系均以钌联吡啶染料分子作为模板客体分子,当ATP存在时,DNA双链结构被打开,孔道中的染料分子得以释放。实验结果显示,两种体系对ATP均呈现出良好的刺激响应性,且采用三螺旋DNA为封堵剂构建的ATP响应控释体系的封堵效果和ATP刺激响应性在同等条件下比采用双链DNA的效果更好,有望应用于药物控释领域。
4.3 核酸适配体-磁性纳米颗粒
磁性纳米颗粒(MNPs)粒径一般为10~100 nm,比表面积大,表面易于修饰,能与特异性配体结合成为靶向载体。MNPs既可通过外加磁场准确靶向并富集在病变部位,降低对正常细胞的毒害,还可在交变磁场作用下吸收电磁波产生热能,同时实现热疗与药物控释。超顺磁氧化铁纳米颗粒(SPION)被认为是最重要的磁性纳米颗粒之一,其生物相容性好,不仅可用作药物载体,而且使用传统的影像技术即可实现肿瘤“可视化”,是“诊疗一体化”技术的关键[76]。
表柔比星(Epi)是一种含蒽环的抗癌药,临床使用表现出剂量依赖型的心脏毒性及骨髓抑制等副作用,这限制了它在治疗癌症方面的应用。Seyed等[77]将Epi与靶向MUC-1的核酸适配体5TR1结合,再与热交联SPION构成Epi-5TR1-SPION三元复合物,相较不表达MUC-1的对照细胞CHO-K1,MUC-1过表达的小鼠结肠癌细胞C26可有效吸收这种三元复合物。将C26细胞接种在小鼠皮下特定区域,磁共振成像显示纳米磁颗粒SPION高度富集在肿瘤细胞区域,活体实验同时表明Epi-5TR1-SPION三元复合物可靶向输运Epi到C26细胞,并有效抑制C26细胞的繁殖,实现了诊疗一体化的治疗模式。
Pala等[78]构建了葡聚糖包裹的MNPs,与特异性识别HER2的DNA核酸适配体结合,用于肿瘤细胞的磁热疗法。实验结果显示,该复合体系可高特异性靶向HER2过表达的SK-BR3细胞,借助磁热治疗可杀死50%的SK-BR3细胞,治疗效果相当于单独MNPs的90倍,且不表达HER2的U-87MG细胞存活率接近100%,大大降低了传统磁热治疗所需MNPs的剂量,进一步降低治疗方法的副作用。
4.4 核酸适配体-单壁碳纳米管
单壁碳纳米管(SWCNTs)是单层石墨片围绕中心按一定的螺旋角卷曲而成的无缝、中空纳米管。由于其独特的理化性质,如细胞膜穿透性能优良、载药量高、pH响应控释、体内循环时间长以及特殊的光学性能等,已成为纳米药物载体的研究热点。
Zhang等[79]用核酸适配体AS1411修饰SWCNTs,并将Dox有效装载到SWCNTs表面构成DOX-AS1411-SWCNTs靶向给药体系。该体系Dox负载量可达121%,能通过pH响应实现药物控释。研究表明,该三元复合体系在2h内能够通过AS1411介导的内吞作用进入细胞,且对食管癌细胞EC-109的抑制效果比单独使用Dox时更强。此外,用808nm近红外光照射可对肿瘤细胞进行光热治疗,证实了DOX-AS1411-SWCNTs具有化疗和热疗协同治疗的效果。
Taghavi等[80]则在修饰的靶向载体AS1411-SWCNT上连接靶向凋亡抑制基因pBcl-xL的shRNA以及抗癌药Dox,形成Dox-pBcl-xLshRNA-SWCNT-AS1411复合物,发现此复合物可以高选择性靶向富含核仁素的胃癌细胞,shRNA可以阻止凋亡抑制基因pBcl-xL的表达,而Dox可以杀死胃癌细胞,所需Dox剂量仅为IC50的1/58,大大降低了Dox的毒副作用,实现基因疗法和化疗的协同功效。这种双重抑制靶向给药体系为临床上安全有效地治疗肿瘤提供了重要的研究基础。
4.5 核酸适配体-脂质体
脂质体(Liposome)是一种人工生物膜,具有两亲性,载药范围广泛,既能封装亲水性药物,也能将疏水性药物负载于磷脂双分子层上[81]。与其他药物载体相比,脂质体具有良好的生物相容性和生物可降解性,尺寸可调,低毒低成本,是良好的药物载体,迄今已有数十种脂质体载药体系被批准用于临床[82]。在多种脂质体的修饰方式中,靶向修饰是提高脂质体递药效率、选择性、细胞内化率,进而提高治疗效果,降低副作用的有效方式。将脂质体与高选择性的核酸适配体相连后形成靶向递药体系,输运药物可以是无机或有机药物小分子,也可以是多肽、蛋白质或者miRNA、siRNA或shRNA等小分子核酸。
Baek等[83]设计了一种新的封装抗癌药物Dox的脂质体。他们通过后嵌入方法(Post-insersion method)将特异性靶向PSMA的RNA核酸适配体嵌入脂质体中,形成90~100 nm的核酸适配体-脂质体复合物,能特异性结合表达PSMA的前列腺上皮细胞LNCaP。将抗癌药物Dox封装于核酸适配体-脂质体内,该复合物对靶细胞LNCaP的毒性增强,杀伤率增大,而非靶细胞的存活率增大。将此复合体系注射入移植LNCaP细胞的裸鼠中,观察到纳米粒富集在肿瘤组织周围,连续注射后,肿瘤有明显缩小现象。用Apt-脂质体封存毒性较强的抗癌药物可以降低临床应用的副作用。
Liao等[84]制备了一种核酸适配体AS1411修饰的新型脂质体,载有DOX和碳酸氢铵(起泡剂),能够克服肿瘤细胞的多重耐药性。AS1411-脂质体对表面过表达核仁蛋白的MCF-7/ADR乳腺癌细胞呈现出高亲和性和特异性结合能力,细胞摄取率明显提高,而未经修饰的脂质体很难进入MCF-7/ADR细胞。经热处理后,包裹在脂质体内的碳酸氢铵分解释放出CO2气体,破坏了磷脂双分子层结构,最终在细胞内部促进DOX的快速释放。此外,体内实验还证明了采用AS1411修饰的脂质体能够显著增加DOX在肿瘤部位的富集量,显著抑制肿瘤细胞生长。
对脂质体等纳米颗粒而言,保证足够的载药率并在靶标附近控制释药速率达到治疗浓度仍是制约纳米载药体系发展的难题。为提高载药率,Plourde等[85]设计了一系列靶向Dox的DNA核酸适配体,发现核酸适配体的长度、结构均会影响核酸适配体的载药性能和释药性能。将Doxapt连接脂质体,将有效驱动药物进入脂质体中,在pH为7.4的PBS溶液中或pH为5的醋酸缓冲液中,脂质体水解,缓慢释放药物。中等长度的Doxapt-30(30nt)同时拥有较高的载药率,可控的释药速率,良好的治疗效果。这项研究显示了在靶向癌细胞的核酸适配体序列中增加靶向药物的序列,可以缔造多功能递药体系。
4.6 核酸适配体-胶束
胶束由两亲性高分子在水中自组装形成,具有独特的壳-核结构,疏水性内核用于装载药物,亲水性外壳则与周围水分子作用形成水化层来稳定胶束。这种特殊结构赋予了胶束作为药物载体的优良性质,如体内循环时间长、疏水性药物在水中溶解度增大、表面易于修饰、药物控释等。
Pluronic F127 (PF)是一种A-B-A型三嵌段共聚物,由聚氧乙烯(A)和聚氧丙烯(B)构成。随着PF水溶液温度升高,聚氧丙烯失水形成疏水内核,而水化的聚氧乙烯则形成亲水外壳,构成胶束。为了增加胶束稳定性,提高药物负载率,Nguyen等[86]在PF中嫁接阳离子型多聚糖——壳聚糖(Chi)形成Chi-TPP-PF胶束,胶束表面羧基化,与靶向HER2的核酸适配体连接,并负载抗癌药物紫杉醇(PTX)。体外细胞实验表明,这种胶束对表达HER2的人乳腺癌细胞杀死率为89%~93%,而对不表达HER2的NV-VN-31细胞的杀死率仅为2%~7%,有效实现了抗癌药物的靶向输运。
Zhang等[87]合成出一种具有双重功能的胶束体系,该体系由pH响应的D-α-生育酚聚乙二醇1000琥珀酸酯(TPGS-b-PBAE,TP)和AS1411核酸适配体(Apt)组成,内部封装有PTX,能够特异性识别表面过表达核仁蛋白的肿瘤细胞。PTX/Apt-混合胶束在pH=7.4的环境中能稳定存在,但在弱酸性环境(pH=5.5) 下会发生解体,迅速释放出内部的PTX。研究结果表明,与无核酸适体修饰的胶束相比,经AS1411修饰的胶束被SKOV3卵巢癌细胞的摄取率更高,富集在肿瘤部位的PTX浓度明显增加,阻碍了肿瘤生长,降低了PTX对正常骨髓细胞的抑制。
4.7 核酸适配体-聚合物纳米粒子
聚乳酸-羟基乙酸共聚物(PLGA)是一种可生物降解的聚合物。PLGA纳米粒生物相容性好,无毒副作用,且具有良好的成囊、成膜性能,受到了广大研究者的青睐。
上皮细胞粘附分子(Epithelial cell adhesion molecule,EpCAM)作为肿瘤干细胞标志物,能在不同的腺癌细胞系中高表达,而在正常细胞中表达水平很低,因而能作为核酸适配体介导的肿瘤治疗的靶标。Li等[88]将PEG化的卵磷脂与PLGA纳米粒子结合用以装载姜黄素,表面则用特异性识别EpCAM蛋白的RNA核酸适配体进行修饰。结果显示,表面有EpCAM表达的结肠癌细胞HT29与纳米粒间的亲和力更强,对纳米粒的摄取率更高,证实了该体系能靶向结肠癌细胞,提高化疗疗效。
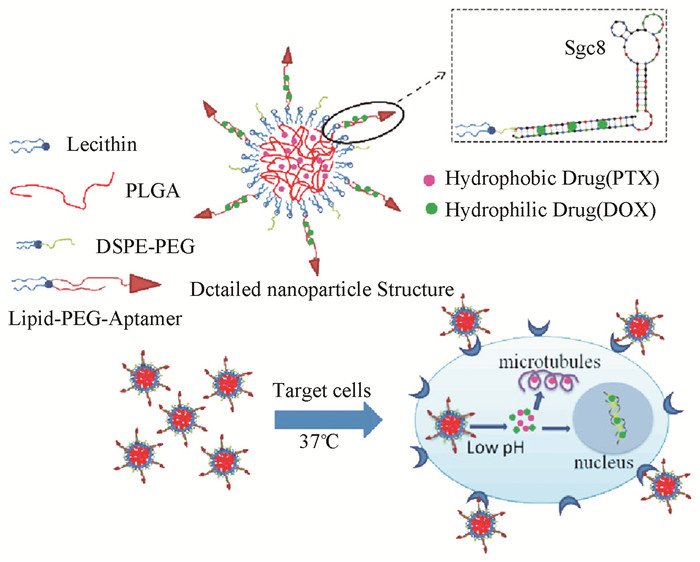
Huang等[89]基于聚合物-脂质体的壳-核结构,构建出核酸适配体sgc8c修饰的PLAG杂化纳米粒子,可同时运输Dox和PTX两种水溶性不同的药物,如图 4所示。实验结果显示,该纳米粒子能被靶细胞CEM细胞选择性摄取,对肿瘤细胞的杀伤作用也大大增强。
Mosafer等[90]用核酸适配体AS1411修饰PLGA纳米粒,同时负载SPION和Dox,SPION/Dox-NPs-AS1411的平均尺寸可达130nm,SPION负载率16%,Dox负载率3%。这种体系不仅能将Dox靶向输运到小鼠结肠癌细胞C26,增加C26细胞对Dox的吸收,高度抑制C26细胞的生长,延长了移植C26结肠癌细胞小鼠的寿命,而且由于磁性纳米粒富集在肿瘤细胞周围,提高了磁共振成像时肿瘤细胞的对比度,具有治疗和诊断的双重功效。
5 结语
尽管核酸适配体已展现出在肿瘤靶向治疗方面的应用潜力,但从实验室走向临床还需克服一系列技术难题。肿瘤的发生发展是一个动态的多因素的复杂过程,培养液内的肿瘤细胞与活体内的肿瘤组织有较大的病理条件的差异,在体外细胞实验甚至在动物模型中表现良好的核酸适配体在病人体内可能会有截然不同的效果。基于组织的SELEX过程(Tissure-based SELEX)和活体SELEX过程(in vivo-SELEX)[91]则在更为接近病理条件的环境中进行核酸适配体的筛选,是未来SELEX过程的发展方向。
另外核酸适配体筛选过程长,成功率低,这也制约了核酸适配体的应用。基于树脂珠的筛选(Bead-based selection)仅需一轮就可以快速筛出核酸适配体,而SOMAmer[92]技术则将核酸适配体的成功率由低于30%提升至超过50%,特别是QPASS(Quantitive parallel aptamer selection system)[93]同时整合了微流控选择、高通量测序、原位合成核酸适配体微阵列等技术,可同时测量成千上万个寡核苷酸的亲和性和特异性,大大加快了筛选的速率。不过这些技术还不够完善,今后还需要进一步发展核酸适配体的快速筛选方法,提高筛选成功率。
再者,尽管化学修饰及PEG修饰能增加核酸适配体在体内的稳定性,但化学修饰较为繁琐,特殊核苷酸的成本较高,PEG修饰尽管简便易行,但已有报道称PEG修饰会诱导产生anti-PEG的抗体[94],因此还需要发展新的成本低、操作简便的稳定核酸适配体的方法。
此外,设计多价或多靶向核酸适配体,增加核酸适配体与靶标的亲和性,可增加输运效率;将核酸适配体同时输运化疗药物和siRNA、miRNA等小分子核酸,实现化疗和基因疗法的协同治疗,是增加药效的方法;将核酸适配体和磁性纳米粒子组成复合体,可实现诊疗一体化的治疗模式。总之,尽管挑战依然存在,但我们相信核酸适配体在未来的肿瘤靶向治疗中将会发挥不可小觑的作用。
-
-
[1]
A D Ellington, J W Szostak. Nature, 1990, 346(6287):818~822. doi: 10.1038/346818a0
-
[2]
C Tuerk, L S Gold. Science, 1990, 249(4968):505~510. doi: 10.1126/science.2200121
-
[3]
J C Cox, A D Ellington. Bioorg. Med. Chem., 2001, 9(10):2525~2531. doi: 10.1016/S0968~0896(01)00028~1
-
[4]
D Eulberg, K Buchner, C Maasch et al. Nucl. Acids Res., 2005, 33(4):e45.
-
[5]
C P Rusconi, E Scardino, J Layzer et al. Nature, 2002, 419(6902):90~94. doi: 10.1038/nature00963
-
[6]
K M Bompiani, D M Monroe, F C Church et al. J. Thromb. Haemost., 2012, 10(5):870~880. doi: 10.1111/j.1538~7836.2012.04679.x
-
[7]
J Soutschek, A Akinc, B Bramlage et al. Nature, 2004, 432(7014):173~178. doi: 10.1038/nature03121
-
[8]
A Nitsche, A Kurth, A Dunkhorst et al. BMC Biotechnol., 2007, 7(1):48. doi: 10.1186/1472~6750~7~48
-
[9]
G Mayer. Angew. Chem. Int. Ed., 2009, 48(15):2672~2689. doi: 10.1002/anie.200804643
-
[10]
X Yan, X Gao, Z Zhang. Genomics Proteomics Bioinformatics, 2004, 2(1):32~42. doi: 10.1016/S1672~0229(04)02005~4
-
[11]
M Kuwahara, N Sugimoto. Molecules, 2010, 15(8):5423~5444. doi: 10.3390/molecules15085423
-
[12]
I Lebars, T Richard, C D Primo et al. Blood Cells Mol. Dis., 2007, 38(3):204~209. doi: 10.1016/j.bcmd.2006.11.008
-
[13]
F J Hernandez, K R Stockdale, L Huang et al. Nucl. Acid Ther., 2012, 22(1):58~68.
-
[14]
N Li, H H Nguyen, M Byrom et al. PLoS One, 2011, 6(6):e20299.
-
[15]
N Derbyshire, S J White, D H Bunka et al. Anal. Chem., 2012, 84(15):6595~6602. doi: 10.1021/ac300815c
-
[16]
L Tan, K G Neoh, E T Kang et al. Macromol. Biosci., 2011, 11(10):1331~1335. doi: 10.1002/mabi.201100173
-
[17]
R M Boomer, S D Lewis, J M Healy et al. Oligonucleotides, 2005, 15(3):183~195. doi: 10.1089/oli.2005.15.183
-
[18]
C P Rusconi, J D Roberts, G A Pitoc et al. Nat. Biotechnol., 2004, 22(11):1423~1428. doi: 10.1038/nbt1023
-
[19]
K Schmidt, S Borkowski, J Kurreck et al. Nucl. Acids Res., 2004, 32(19):5757~5765. doi: 10.1093/nar/gkh862
-
[20]
D Eulberg, S Klussmann. ChemBioChem., 2003, 4(10):979~983. doi: 10.1002/cbic.200300663
-
[21]
J J Turner, J S Hoos, S Vonhoff et al. Nucl. Acids Res., 2011, 39(21):e147.
-
[22]
W Sun, L Du, M Li. Curr. Pharm. Des., 2011, 17(1):80~91. doi: 10.2174/138161211795049769
-
[23]
M Ye, J Hu, M Peng et al. Int. J. Mol. Sci., 2012, 13(3):3341~3353.
-
[24]
J Mi, Y Liu, Z N Rabbani et al. Nat. Chem. Biol., 2010, 6(1):22~24. doi: 10.1038/nchembio.277
-
[25]
H U Göringer. Trends Parasitol., 2012, 28(3):106~113. doi: 10.1016/j.pt.2011.12.005
-
[26]
M Moreno, V M González. Curr. Med. Chem., 2011, 18(32):5003~5010. doi: 10.2174/092986711797535218
-
[27]
J C Gilbert, T De Feo~Fraulini, R M Hutabarat et al. Circulation, 2007, 116(23):2678~2686. doi: 10.1161/CIRCULATIONAHA.107.724864
-
[28]
J L Diener, H A Lagasse, D Duerschmied et al. J. Thromb. Haemost., 2009, 7(7):1155~1162. doi: 10.1111/j.1538~7836.2009.03459.x
-
[29]
J Bradley, M Ju, G S Robinson. Angiogenesis, 2007, 10(2):141~148. doi: 10.1007/s10456~007~9069~x
-
[30]
L S Green, D Jellinek, R Jenison et al. Biochemistry, 1996, 35(45):14413~14424. doi: 10.1021/bi961544+
-
[31]
A P Mann, T Tanaka, A Somasunderam et al. Adv. Mater., 2011, 23(36):278~282. doi: 10.1002/adma.201101541
-
[32]
A P Mann, R Bhavane, A Somasunderam et al. Oncotarget, 2011, 2(4):298~304. doi: 10.18632/oncotarget.261
-
[33]
J Ruckman, L S Green, J Beeson et al. J. Biol. Chem., 1998, 273(32):20556~20567. doi: 10.1074/jbc.273.32.20556
-
[34]
N Ferrara, H P Gerber, J Lecouter. Nat. Med., 2003, 9(6):669~676. doi: 10.1038/nm0603~669
-
[35]
D Huang, D Vu, L A Cassiday et al. PNAS, 2003, 100(16):9268~9273. doi: 10.1073/pnas.1632011100
-
[36]
L L Lebruska, M L Rd. Biochemistry, 1999, 38(10):3168~3174. doi: 10.1021/bi982515x
-
[37]
B J Hicke, C Marion, Y F Chang et al. J. Biol. Chem., 2001, 276(52):48644~48654. doi: 10.1074/jbc.M104651200
-
[38]
K S Schmidt, S Borkowski, J Kurreck et al. Nucl. Acids Res., 2004, 32(19):5757~5765. doi: 10.1093/nar/gkh862
-
[39]
S E Lupold, B Hicke, Y Lin et al. Cancer Res., 2002, 62(14):4029~4033.
-
[40]
O C Farokhzad, S Jon, A Khademhosseini et al. Cancer Res., 2004, 64(21):7668~7672. doi: 10.1158/0008~5472.CAN~04~2550
-
[41]
Y Song, Z Zhu, Y An et al. Anal. Chem., 2013, 85(8):4141~4149. doi: 10.1021/ac400366b
-
[42]
S Shigdar, J Lin, Y Yu et al. Cancer Sci., 2011, 102(5):991~998. doi: 10.1111/j.1349~7006.2011.01897.x
-
[43]
S D Gomes, J Miguel, L Azema et al. Bioconjug. Chem., 2012, 23(11):2192~2200. doi: 10.1021/bc300146c
-
[44]
B Hicke, A W Stephens, T A Gould et al. J. Nucl. Med., 2006, 47(4):668~678.
-
[45]
H Shi, X He, K Wang et al. PNAS, 2011, 108(10):3900~3905. doi: 10.1073/pnas.1016197108
-
[46]
A Nolte, S Klussmann, R Bald et al. Nat. Biotechnol., 1996, 14(9):1116~1119. doi: 10.1038/nbt0996~1116
-
[47]
S G Sayyed, H Hagele, O P Kulkarni et al. Diabetologia, 2009, 52(11):2445~2454. doi: 10.1007/s00125~009~1493~6
-
[48]
E Ng, A P Adamis. Ann. Ny. Acad. Sci., 2006, 1082(1):151~171. doi: 10.1196/annals.1348.062
-
[49]
P Celec, Y Yonemitsu. Pathophysiology, 2004, 11(2):69~75. doi: 10.1016/j.pathophys.2004.03.002
-
[50]
J Lee, M D Canny, A D Erkenez et al. PNAS, 2005, 102(52):18902~18907. doi: 10.1073/pnas.0509069102
-
[51]
Eyetech Study Group. Retina, 2002, 22(2):143~152. doi: 10.1097/00006982~200204000~00002
-
[52]
J Huang, J T Moore, S Z Soffer et al. J. Pediatr. Surg., 2001, 36(2):357~361. doi: 10.1053/jpsu.2001.20716
-
[53]
A C Girvan, Y Teng, L K Casson et al. Mol. Cancer Ther., 2006, 5(7):1790~1799. doi: 10.1158/1535~7163.MCT~05~0361
-
[54]
S Soundararajan, W Chen, E K Spicer et al. Cancer Res., 2008, 68(7):2358~2365. doi: 10.1158/0008~5472.CAN~07~5723
-
[55]
E M Reyesreyes, F R Salipur, M Shams et al. Mol. Oncol., 2015, 9(7):1392~1405. doi: 10.1016/j.molonc.2015.03.012
-
[56]
P J Bates, D A Laber, D M Miller et al. Exp. Mol. Pathol., 2009, 86(3):151~164. doi: 10.1016/j.yexmp.2009.01.004
-
[57]
C Ritchie, B Doran, K Shah et al. Proc. Am. Assoc. Cancer Res., 2007, 48:4818.
-
[58]
D T Fearon. Cancer Immunol. Res., 2014, 2(3):187~193. doi: 10.1158/2326~6066.CIR~14~0002
-
[59]
刘珍宝, 石依倩, 陈长仁等. 科学通报, 2014, 59(14):1267~1279. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=kxtb201414001&dbname=CJFD&dbcode=CJFQ
-
[60]
V Bagalkot, O C Farokhzad, R Langer et al. Angew. Chem., 2006, 118(48):8329~8332. doi: 10.1002/ange.200602251
-
[61]
Y Hu, J Duan, Q Zhan et al. PLoS ONE, 2012, 7(2):e31970.
-
[62]
Z Liu, J Duan, Y Song et al. J. Transl. Med., 2012, 10(1):148. doi: 10.1186/1479~5876~10~148
-
[63]
Y Huang, D Shangguan, H Liu et al. ChemBioChem, 2009, 10(5):862~868. doi: 10.1002/cbic.200800805
-
[64]
G Zhu, J Zheng, E Song et al. PNAS, 2013, 110(20):7998~8003. doi: 10.1073/pnas.1220817110
-
[65]
P Mallikaratchy, Z Tang, W Tan et al. ChemMedChem, 2008, 3(3):425~428. doi: 10.1002/cmdc.200700260
-
[66]
S H Lee, Y Y Kang, H E Jang et al. Adv. Drug Deliver. Rev., 2016(104), 78~92
-
[67]
J O McNamara, E R Andrechek, Y Wang et al. Nat. Biotechnol., 2006, 24(8):1005~1015. doi: 10.1038/nbt1223
-
[68]
J Zhou, H Li, J Zhang et al. J. Vis. Exp., 2011, 52(52):e2954.
-
[69]
H Yoo, H Jung, S A Kim et al. Chem. Commun., 2014, 50(51):6765~6767. doi: 10.1039/c4cc01620c
-
[70]
唐德平, 毛爱红, 王芳等. 中国生物工程杂志, 2015, 35(1):54~60. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=swgj201501008&dbname=CJFD&dbcode=CJFQ
-
[71]
Y Huang, K Sefah, S Bamrungsap et al. Langmuir, 2008, 24(20):11860~11865. doi: 10.1021/la801969c
-
[72]
Y Luo, Y Shiao, Y Huang et al. ACS Nano, 2011, 5(10):7796~7804. doi: 10.1021/nn201592s
-
[73]
Y Guo, S Li, J Liu et al. Sens. Actuat. B, 2016,235:655~662. doi: 10.1016/j.snb.2016.05.131
-
[74]
X Yang, X Y Liu, Z Liu et al. Adv. Mater., 2012, 24(21):2890~2895. doi: 10.1002/adma.201104797
-
[75]
赵应香. 湖南大学硕士学位论文, 2013.
-
[76]
R Tietze, S Lyer, S Dürr et al. Nanomedicine, 2012, 7(3):447~457. doi: 10.2217/nnm.12.10
-
[77]
S H Jalalian, S M Taghdisi, N Shamedani. Eur. J. Pharm. Sci., 2013, 50(2):191~197. doi: 10.1016/j.ejps.2013.06.015
-
[78]
K Pala, A Serwotka, F Jelen et al. Int. J. Nanomed., 2014, 9(1):67~76.
-
[79]
H Zhang, C Chen, L Hou et al. J. Drug Target., 2013, 21(3):312~319. doi: 10.3109/1061186X.2012.749880
-
[80]
S Taghavi, A H Nia, K Abnous et al. Int. J. Pharmaceut., 2017, 516(1~2):301~312. doi: 10.1016/j.ijpharm.2016.11.027
-
[81]
H Daraee, A Etemadi, M Kouhi et al. Artif. Cells Nanomed. Biotechnol., 2016, 44(1):381~391. doi: 10.3109/21691401.2014.953633
-
[82]
H Bardania, S Tarvirdipour, F Dorkoosh, Artif. Cells Nanomed. Biotechnol., 2017, 45:1~12.
-
[83]
S E Baek, K H Lee, Y S Park et al. J. Control. Release, 2014, 196:234~242. doi: 10.1016/j.jconrel.2014.10.018
-
[84]
Z X Liao, E Y Chuang, C C Lin et al. J. Control. Release, 2015, 208(28):42~51.
-
[85]
K Plourde, R M Derbali, A Desrosier et al. J. Control. Release, 2017, 251:82~91. doi: 10.1016/j.jconrel.2017.02.026
-
[86]
K T Nguyen, T H Nguyen, D H Do et al. Adv. Nat. Sci.:Nanosci. Nanotechnol., 2017, 8(1):5002~5009.
-
[87]
J Zhang, R Chen, X Fang et al. Nano Res., 2015, 8(1):201~218. doi: 10.1007/s12274~014~0619~4
-
[88]
L Li, D Xiang, S Shigdar et al. Int. J. Nanomed., 2014, 21(9):1083~1096.
-
[89]
F Huang, M You, T Chen et al. Chem. Commun., 2014, 50(23):3103~3105. doi: 10.1039/c3cc49003c
-
[90]
J Mosafer, K Abnous, M Tafaghodi et al. Eur. J. Pharm. Sci., 2017,113:60~74.
-
[91]
J Mi, Y M Liu, Z N Rabbani. Nat. Chem. Biol., 2010, 6:22~24. doi: 10.1038/nchembio.277
-
[92]
S Kraemer, J D Vaught, C Bock et al. PLoS One, 2011, 6(10):e26332.
-
[93]
M Cho, S S Oh, J Nie et al. PNAS, 2013,110(46):18460~18465. doi: 10.1073/pnas.1315866110
-
[94]
M G P Saifer, L D Williams, M A Sobczyk et al. Mol. Immunol., 2014,57(2):236~246. doi: 10.1016/j.molimm.2013.07.014
-
[1]
-


-


 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 401
- 文章访问数: 19883
- HTML全文浏览量: 7762
 下载:
下载:
