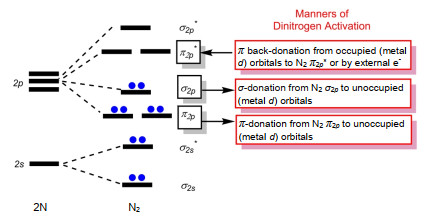
Figure 1.
Schematic MO diagram involving dinitrogen activation.
Recent Developments of Dinitrogen Activation on Metal Complexes and Clusters
Xue-Lu Ma , Meng Li , Jun-Bo Lu , Cong-Qiao Xu , Jun Li

Dinitrogen is the most abundant substance in the atmosphere, and its activation is a critical process for the life cycle on the earth. The transformations of N2 into NH3 and organonitrogen species are indispensable and essential for life. However, the process for cleavage and conversion of dinitrogen has been identified as a significant challenge.[1] The high bond dissociation energy of the N≡N triple bond partly accounts for the thermodynamical difficulty. Lots of factors hindering dinitrogen activation mostly involve kinetics in nature. While the large HOMO-LUMO (the highest occupied molecular orbital – the lowest unoccupied molecular orbital) gap disfavors electron transfer, the low proton affinity makes direct protonation of dinitrogen extremely difficult under ambient conditions. Indeed, the high ionization potential, negative electron affinity, and non-polar strong bonds give rise to the inertness and low reactivity.[2, 3]
Qualitative Mulliken-Hund molecular orbital (MO) theory renders the familiar (σs)2(σ2s*)2(π2p)4(σ2p)2(π2p*)0(σ2p*)0 ground-state electron configuration for triple-bonded N2. Inspired by Dewar-Chatt-Duncanson (DCD) σ-donor/π-acceptor bonding model for alkene, [4] dinitrogen activation can be classified into three categories as shown in Figure 1. Firstly, the lowest unoccupied molecular orbital (LUMO, π2p*) of N2 can act as a Lewis acid site to accept electron density from an occupied metal d-orbital to form a π back-bond, or from external electrons undergoing a reduction reaction. Secondly, N2 is able to bind as a Lewis base and donate electron density from the HOMO (σ2p) into an unoccupied metal d-orbital, which results in the formation of a σ-type bond. Thirdly, the π2p orbital of N2 could also donate electron density to unoccupied metal d-orbitals, thus weakening the N≡N bond. In addition, the mixture of the above three ways to reduce or 'activate' the N≡N bond could be fulfilled in multi-metallic cluster systems. The increase of the N-N bond length as well as the decrease of the N-N bond order, force constant, and stretching frequency are used to identify weakening of the N-N bond.[5]
In general, dinitrogen activation on metal-based complexes and clusters can proceed either via direct dissociative pathway or indirect associative pathway.[5] In the direct dissociative activation, the N≡N unit is directly dissociated into two N atoms, which is typically seen in systems with multiple active sites or strongly reducing early transition metals. In the associative activation, the N≡N triple bond is gradually weakened accompanied with the functionalization of N atoms by other agents (e.g. proton/electron, hydrogen, halides, etc.).[6, 7]
The industrial Haber-Bosch process converts N2 and H2 into NH3 on Fe-or Ru-based heterogeneous metal catalysts under high-temperature and high-pressure operating conditions. The well-studied atomistic mechanism of the process is verified to be a direct N≡N dissociation process on multi-metallic active sites, involving the so-called C7-site of Fe(111) or Fe(211) surface or the B5-site of Ru(0001) step, which is the rate-determining step.[8, 9] However, biological nitrogen fixation occurs under ambient conditions, which is accomplished by multi-metallic clusters in the nitrogenase cofactors via the associative dinitrogen activation.[10] Compared to the multifactorial active sites in the industrial and biological nitrogen fixation, it is instructive to perform mechanistic investigations on dinitrogen activation by model metal complexes and clusters using well-defined active sites.
Over the last half century, a series of metal complexes and clusters binding with dinitrogen were synthesized and identified, involving mono-nuclear or multi-nuclear metal active sites. This work is to provide an overview on mechanistic aspects of dinitrogen activation. We will briefly discuss the N2 binding mode associated with the dinitrogen activation, and then discuss selected dinitrogen complexes based on different coordination binding modes. Representative examples and recent progress of dinitrogen activation on metal clusters will also be provided. At last, a perspective will be given on potential challenges in this field.
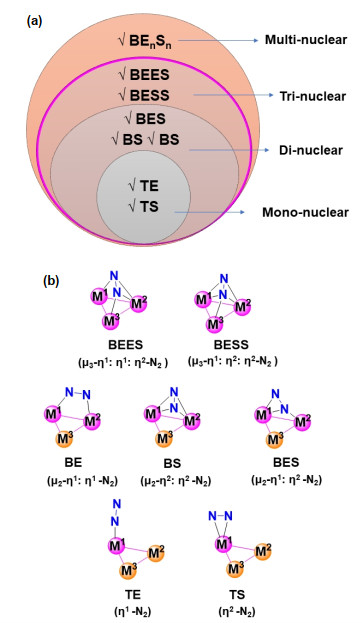
The dinitrogen coordination mode plays an essential role in dinitrogen activation.[11] Comprehensive experimental and theoretical studies have demonstrated that dinitrogen can coordinate with the metal centers in terminal or bridging manners as summarized in Figure 2(a). Following the convention in coordination chemistry, when dinitrogen bridges metal sites, it is normally denoted as μn, where n is the number of bridged metals (μ2 is usually abbreviated as μ). Based on the interaction between dinitrogen and each metal center, binding mode can be distinguished into "end-on" and "side-on", which are represented as η1 (one N-atom bounds to the metal) and η2 (two N-atoms bound to the metal), respectively.[10] For terminal dinitrogen bounded to a mononuclear transition metal, the side-on mode was found only in late transition metal complexes, while the end-on coordination mode was seen in mostly all existing mononuclear transition metal complexes.[12] In multi-nuclear complexes or clusters, the binding mode of N2 is richer, which is the combination of various active sites, as exemplified by the binding mode of N2 in trinuclear complexes or clusters shown in Figure 2(b).
In weakly activated dinitrogen, one of the most typical bonding modes is mononuclear terminal end-on binding, which can be explained in a manner entailing σ-donation from the dinitrogen moiety to the metal, as well as some π back-bonding from the metal d orbitals to π*-orbitals of dinitrogen. It is revealed that σ-donation is more crucial in this binding mode, while back-bonding plays a weaker role due to long M···N2 distance.[13] Based on this generalization, the coordinated dinitrogen is best referred to as formally neutral and illustrated as (N2)0. Once the dinitrogen is strongly polarized, the terminal nitrogen is endowed with a formally negative charge, so that electrophilic attack by other agents can easily activate the unit of dinitrogen.[5]
In order to strongly activate dinitrogen, a powerful reducing metal center is indispensable, which is most typically observed with the early transition metals, especially with low-valent ones.[14] As the result of strongly reducing nature of the active metals with the availability of high-lying d-orbital electrons for π-back donation, the dinitrogen can be considered as (N2)δ- (δ = -1 to -4), which could be simply figured out by N-N stretching frequency and the N-N bond length. Additionally, most of compounds with strongly activated dinitrogen are not mononuclear, but the multi-nuclear cluster systems. Especially, each metal in dinuclear complexes utilizes two electrons to back-donate to the N2 unit, leaving one of the N2 π*-orbitals complimentary to engage with another metal center, which facilitates the formation of dinuclear dinitrogen complex.[15] Therefore, the metal oxidation state and coordination number greatly influence the activation of dinitrogen.[16]
Additionally, the peripheral ligands of the metal site also play a significant role in determining the extent of N-N activation. Large differences in reactivity can arise from subtle modifications in ligand electronic structure, steric geometry, and bite angle of peripheral ligands, among others.[17] Typically, the geometric modifications are implemented using bulky peripheral ligands.[18] For example, the addition of methyl groups to cyclopentadienyl ligands of bis(cyclopentadienyl)M (M = Zr, Hf) dimer affects the activation of dinitrogen, usually by forcing the two metal atoms apart to change the binding mode.[19] Especially, a series of studies on hindered β-diketiminate complexes has revealed that a trigonal planar geometry at the metal results in the weakness of N-N bond on iron, cobalt, and nickel[20] though these metals do not usually have sufficient back-bonding capacity to reduce dinitrogen. Similarly, the N-N bond activation can also be achieved by tetrahedral ligands, [21] which induces strong π-back-bonding as explained by crystal-field analysis.[22]
Moreover, the modifications of late transition-metal orbital energies caused by peripheral ligands with proper electron-withdrawing substituent groups can also promote the changes of N-N bond order in dinitrogen activation.[23] The tri-nucleating macrobicyclic ligands could cause metal σ- and π-type frontier orbitals oriented toward the internal cavity of the cyclophane, providing the premise conditions for dinitrogen activation.[24] In 2021, Murray and coworkers synthesized a 48-electron Co3I cluster with mono-valent CoI, Co3LEt/Me, in which electrons between the metal-metal bonding interaction assist in the cooperative activation of dinitrogen, and Co3N2LEt/Me is formed with a μ3-η1: η1: η2 coordination mode.[25] Therefore, it is an effective method to utilize the geometric and electronic manipulation around a metal active center to modulate the extent of back-bonding and dinitrogen activation.
As the first characterized N2 complex, [Ru(NH3)5(N2)]2+ was reported with the discovery of terminal end-on N2 binding (η1-N2) in 1965.[26] In this bound mode, the dinitrogen atoms and metal center are essentially collinear, which turns out to be most commonly observed for N2. A number of different coordination modes of N2 have since then be found, including the bridging end-on mode of μ-η1: η1-N2 diruthenium complex in 1968.[27] The first bridging side-on N2 complex, [Sm(C5Me5)2]2(μ-η2: η2-N2), was isolated in 1988, and asymmetric bridging end-on-side-on mode was realized in the dinitrogen complex [(NPN)Ta(μ-H)]2(μ-η1: η2-N2), where NPN is PhP(CH2SiMe2NPh)2.[28, 29] However, the major drawback of many of these systems was that it was difficult to separate the compounds from reaction products for the catalytic reuse.[30] In 2003, a groundbreaking homogeneous catalytic ammonia production system was achieved using a well-defined tri(amido)amine Mo(Ⅲ) complex reported by Schrock and coworkers.[31] In 2011, the low-valent Mo-phosphine catalyst for ammonia production was reported by Nishibayashi and coworkers.[32] In 2013, it was reported by Peters and coworkers that a tris(phosphine)borane iron complex was capable of binding and reducing dinitrogen to produce ammonia.[33]
Significant progresses have been made in developing metal complexes for activating dinitrogen and reducing dinitrogen into ammonia. Based on both historical and recent studies, we summarize hereby various metal complexes that have been explored for dinitrogen activation and categorize these dinitrogen metal complexes according to their different N2 binding modes, and discuss the dinitrogen activation and functionalization mechanism accordingly.
We will begin with the terminal end-on mode, as shown in Figure 3(a). In this coordination mode, the distal N atom of the coordinated dinitrogen is nucleophilic and may react with various electrophiles.[34, 35] Typically, the electrophile is firstly added to the distal N atom, and the difunctionalized dinitrogen units can be generated by following a second addition to the same distal N atom or to the coordinated N atom.[36] The dinitrogen unit in the terminal end-on N2 complexes can undergo electrophilic functionalization to form ammonia, hydrazine or tris(trimethylsilyl)-amine.[37] A recent remarkable progress by the Peters group shows that low valent, trigonal Co and Fe complexes with terminal end-on N2, [21, 38] can act as precursors for N2 functionalization to catalytic nitrogen fixation.[39, 40] Depending on the various metals and its supporting apical ligand, degrees of N2 activation make a difference based on the N-N stretching vibrational frequency ν(N-N). The effective ligands for facilitating N2 activation are continuously been developed, including the cyclic alkyl amino carbene-based ligands[41] and carbazole-based PNP-type pincer ligands.[42]
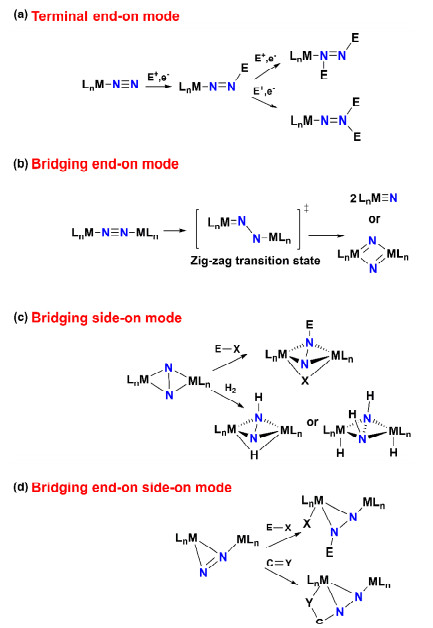
In the bridging end-on mode of dinitrogen (Figure 3(b)), the electron densities of the two bridging N atoms are both reduced through coordination with metal atoms.[43] However, further reduction to the formed metal nitride species is a productive route for the subsequent reaction, [44] since the metal nitrides are easily attacked by electrophiles and nucleophiles. As shown in Figure 3(b), it is derived by the addition of electrons and cleavage of N-N bond involving the nitride formation and the bridging end-on mode can cause the formation of higher value organonitrogen derivatives.[45] For example, bridging end-on N2 complexes of group-7 metals can generate nitrides, with transformation of a (tBu2PCH2CH2)2N- pincer-ligated dinitrogen dirhenium(Ⅱ) complex into two Re(Ⅴ) nitrides.[44] The formation of nitrides was found in the group 5 metal dinuclear N2 complex, [Nb(P2N2)]2(μ-N2) (P2N2 = PhP(CH2SiMe2NSiMe2CH2)2PPh), which can be considered as two Nb(Ⅳ) centers bound with a N24- hydrazido ligand.[46] In general, bridging end-on N2 coordination to low-valent metals with the appropriate ligand design can ultimately give rise to nitrides.
The first bridging side-on N2 complex is the disamarium species [Sm(C5Me5)2]2(μ-η2: η2-N2), [47] in which N-N bond remains nearly unperturbed from free dinitrogen. Generally, the bridging side-on binding mode is usually found in lanthanides, actinides and early transition metals with the elongated N-N bond distance.[48, 49] Moreover, the side-on binding on alkali metals has been proved to enhance the back-donation by stabilizing the N2 unit with negative charge.[50] N2 hydrogenation and functionalization available to the bridging side-on N2 coordination are diverse as displayed in Figure 3(c). For example, the hydrogenation of [Zr(P2N2)](μ-η2: η2-N2) produces the diazenido complex with a bridging hydrido ligand and an N-H bond.[51] The corresponding generation of N-C and N-Si bonds undergoes the heterolytic bond cleavage with terminal arylalkynes and silanes, respectively.[11] With the bridging side-on binding mode, a multimetallic uranium nitride complex is able to convert dinitrogen to ammonia and cyanate.[52]
The widely studied end-on-side-on N2 complex, [Ta(NPN)]-(μ-η1: η2-N2)(μ-H)2 (NPN = PhP(CH2SiMe2NPh)2), forms from the tetrahydride [Ta(NPN)]2(μ-H)4 and N2.[53] In the mode of the end-on-side-on, the geometry and charge distribution of N24- unit are asymmetric with an activated N-N bond.[54] The resulting reactivity of functionalization has shown to be extremely rich, especially for the electrophilic attack at the terminal N atom.[55] The charge localization afforded by end-on-side-on N24- binding provides different opportunities for bond formation with N atoms. Typically, the terminal N atom exhibits nucleophilic character, giving adducts with Lewis acids or bonding with main-group and C-centered electrophiles according to the mechanism as shown in Figure 3(d).
Furthermore, both theoretical and experimental studies indicated that extra metal centers promote dinitrogen activation.[56, 57] Particularly, low-coordinate complexes[58] with three Fe atoms showed multi-metallic cooperation makes N-N bond cleavage thermodynamically feasible.[59] Holland and coworkers developed a series of multinuclear iron complexes to mimic the nitrogenase, [60] and revealed that three-active iron(Ⅰ) centers can engage with dinitrogen in both end-on/side-on/side-on (ESS) and end-on/end-on/side-on (EES) binding modes. The ESS binding mode can easily break the N-N bond without significant structural transformation, while the intermediate with the cleavage N-N bond is not thermodynamically stable. However, the negative charge on the two-coordinate nitride can be stabilized by a positively charged ion.[61] Multi-metallic uranium-rhodium complex can be also used to initiate the cleavage of dinitrogen in the presence of KC8.[62] In the dinitrogen cleavage product, the uranium and rhodium adapted to the +Ⅳ and -I oxidation states, respectively. Jori and coworkers reported the cleavage of dinitrogen to nitrides by a uranium-potassium complex, in which the U(Ⅳ)/U(Ⅵ) tetrauranium cluster generates from successive one-electron transfer, resulting in N2 cleavage and the formation of putative diuranium(Ⅴ) bis-nitride. Noteworthily, cooperative potassium binding to the U-bound N24- ligand facilitates dinitrogen cleavage during electron transfer.[63] Followed by the "hydride electron reservoir" paradigm, Hou and coworkers suggested that the titanium/ruthenium hetero-multi-metallic hydride complexes can serve as a platform for dinitrogen activation.[64]
Metal clusters can be stabilized using peripheral ligands as discussed above. While most experimentally isolated metal clusters involving ligands, gas-phase clusters can provide model systems in understanding the fundamental N2 activation and conversion mechanism. By using matrix isolation techniques, the bare metal clusters can be formed with the aid of thermal evaporation and laser-ablation technique.[65, 66] However, thermal evaporation is more suitable to study dinitrogen activation, which could avoid the dinitrogen cleavage prior to the interaction with the active centers.[67]
Through the combination of experimental and theoretical studies, the mechanisms of metal cluster activation and cleavage of dinitrogen were extensively studied.[68-72] The ligand-free Ti dimer can react with N2 to give the cyclic bis-nitrido species Ti(μ-N)2Ti without any significant activation barrier in just one step, in which the N≡N triple bond is completely cleaved.[73] In reactions of N2 with metal dimers, the interaction with N2 in the end-on-side-on M2(μ2-η2: η1-N2) and bridging side-on M2(μ2-η2: η2-N2) binding modes are revealed as two vital intermediates, in which the N≡N triple bond is significantly activated by the diatomic clusters Gd2[74] and Sc2.[75] The activation mechanism of N2 on bare clusters M2 (M = Gd, Sc, ···) provides important insights for understanding the mechanism of how N2 is activated on low-valent metal clusters and surface and will be discussed in more details later.
Recently, He and coworkers reported a series of studies on dinitrogen activation by gas-phase multi-nuclear metal clusters, such as dinuclear carbide cluster anions Ta2C4-, [69] and the trinuclear nitride cluster anions Ta3N3H0, 1-[68]. Additionally, N2 binds to three metal atoms in the end-on/side-on/side-on (ESS) mode, and M3(μ3-η1: η2: η2-N2) is suggested in the feasible N≡N bond cleavage by trinuclear vanadium carbide cluster anions V3C4-.[71] Dinitrogen binds molecularly to the iron sites of Fe5S2, 3- in a common end-on coordination mode via an unprecedented back-donation interaction from the localized d-d bonding orbitals of Fe-Fe sites to the π* antibonding orbitals of N2.[76] The outstanding reactivity of heteronuclear metal cluster (FeV2C2-) toward N2 benefits from the complementary cooperation of Fe and V atoms in the light of the geometric flexibility of the Fe-V-V ring as well as the electron-withdrawing and electron-donating capacity.[77]
From dinitrogen activation on heterogeneously surface-supported metal clusters, we recently proposed that the singly dispersed bimetallic M1An catalyst serves as a new surface single-cluster catalyst (SCC) for the biomimetic N2-to-NH3 thermal conversion.[78] It is demonstrated that with isolated Rh1Co3 cluster on CoO(011) surface the preferred pathway indeed follows the analogous enzyme-catalyzed associative mechanism, in which a high degree of N2 activation is embodied in a bridging manner as shown in Figure 4(a), as well as the co-adsorption of N2 and H2 is followed by the alternating hydrogenation. The catalytic ability of M1An catalyst arises from both the electronic effect of doped low-valent metal M that serves as a charge buffer and the complementary role of synergic metal atom A in catalysis. Similarly, the small Fe3 clusters supported by θ-Al2O3(010) surface were found theoretically to undergo efficient N2 activation as shown in Figure 4(b).[79] The inert support has little electronic interaction with the Fe3 cluster, and thus maintains Fe in an even more reduced state for metal-metal bonded Fe clusters.[80] In this system, N2 is firstly activated on the Fe3 active center in an EES manner. The key role of multiple active sites in dinitrogen activation has been summarized in Figure 4(c-l).[81-91]
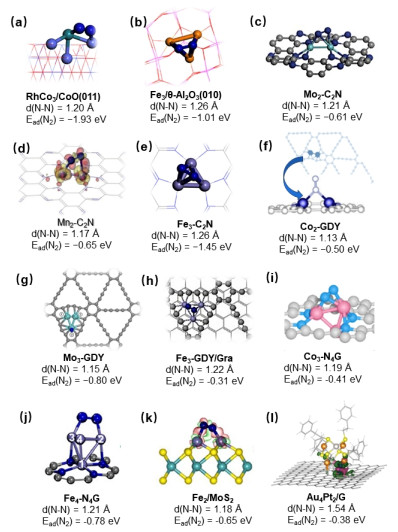
Very recently, Luo and coworkers have demonstrated that graphdiyne supported TiCo3@GDY also exhibits high activities via synergy effect, where the Co3 cluster acts as the electron donor and the heteroatom serves as the single active site throughout the NRR process.[92] Chen and coworkers have reported the ternary ruthenium complex hydrides Li4RuH6 and Ba2RuH6 as an alternative class of catalysts, composed of electron- and hydrogen-rich [RuH6] anionic centers, for non-dissociative dinitrogen reduction.[93, 94] Dai and coworkers have designed an SCC of the Fe4 cluster anchored to 2D GaS with the remarkable NRR performance, in which N2 is completely activated in a side-on adsorption configuration. Therefore, multi-metallic clusters, which can be capable of performing collaborative reduction of the dinitrogen, provide an alternative strategy for the cleavage and transformations of dinitrogen, as well as pave the way for its future application in catalytic processes.[95, 96]
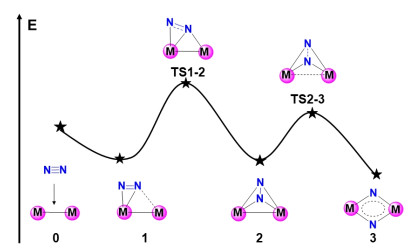
As the simplest and effective model for multi-nuclear active sites, dinuclear metal system could provide insights into the synergistic mechanism of electron rearrangements in dinitrogen activation. The strong interaction between the active centers and dinitrogen mainly results from the π back-donation from the metal centers to N2. The triple bond of dinitrogen is weakened to a certain extent, in which electron density relocates from the metal-metal π bonding to the vacant antibonding orbitals of dinitrogen, and relatively weak donation of electron density from π bonds in dinitrogen to the vacant metal-metal empty orbitals could occur. From the view of geometric transformation, the approaching dinitrogen usually lies in the same plane as dinuclear metals, and end-on-side-on N2 bonded manner 1 is favorable in the following dinitrogen activation as shown in Figure 5, which could be identified in structurally properly characterized systems.[74, 75] Along with the N-N bond distance elongated, the preferred isomer 2 with a nonplanar bridged-bonded C2v symmetry could be rearranged from 1. The end-on-side-on N2 bonded model transfers to a double side-on (μ-η2: η2-N2) "butterfly" geometry. Finally, a planar cyclic [M(μ-N)2M] isomer 3 with a completely cleaved N-N bond is trapped in the deep potential, exhibiting a slightly distorted planar square with D2h symmetry. The process is accompanied by both partial donation of bonding electrons from N2 to metal centers and π-back-donation from the metal centers to the antibonding π* and σ* orbitals of N2. It is suggested that interplay of occupied and empty d-orbitals at metal center dictates the reactivity of the dinuclear metal system with dinitrogen.[74, 75]
In this minireview, we have summarized the recent progresses of dinitrogen activation on metal complexes and clusters, including the species with peripheral ligands, bare metal clusters and surface single-cluster catalysts. As enzymatic active sites in nitrogenase rely upon all metal centers and the surrounding residues to work, the key design principles of redox versatility and cooperative dinitrogen activation by multiple proximal metal centers may be maintained in the metal complexes or clusters. In addition, dinitrogen coordination mode, metal oxidation state, coordination number, and peripheral ligands greatly influence the activation of dinitrogen.
Especially noteworthy is that the electronic coupling of the metal centers might trigger more accessible redox states than those of the individual component metals, [97] and the rich redox character of a metal-metal bonded species might take part in cooperative interactions with dinitrogen. When metal remains in low oxidation state, the metal d-orbital energies are raised and the active site is more effective in reduction reaction. Therefore, it appears that the use of low-valent and multi-metallic systems, which are capable of performing cooperative activation of the dinitrogen, may provide a significant strategy for activating inert molecules like dinitrogen. In metal complexes and metal clusters, the steric factors provide the necessary quantum confinement effect, while the active sites offer selective redox ability for dinitrogen activation and subsequent conversion. Combination of robust support and highly active metal single-atom or single-clusters (including p-, d-, and even f-block metal clusters) may provide highly selective and effective catalysts for atomically precise heterogeneous catalysis.[97, 98]
Himmel, H. J.; Reiher, M. Intrinsic dinitrogen activation at bare metal atoms. Angew. Chem. Int. Ed. 2006, 45, 6264-6288. doi: 10.1002/anie.200502892
Zhan, C. G.; Nichols, J. A.; Dixon, D. A. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J. Phys. Chem. A 2003, 107, 4184-4195. doi: 10.1021/jp0225774
Hong, Q. S.; Li, T. Y.; Zheng, S. S.; Chen, H. B.; Chu, H. H.; Xu, K. D.; Li, S. N.; Mei, Z. W.; Zhao, Q. H.; Ren, W. J.; Zhao, W. G.; Pan, P. Tuning double layer structure of WO3 nanobelt for promoting the electrochemical nitrogen reduction reaction in water. Chin. J. Struct. Chem. 2021, 40, 519-526.
Stahl, S. S. Organotransition metal chemistry: from bonding to catalysis. J. Am. Chem. Soc. 2010, 132, 8524-8525.
Liu, H. M.; Wei, L.; Liu, F.; Pei, Z. X.; Shi, J.; Wang, Z. J.; He, D.; Chen, Y. Homogeneous, heterogeneous, and biological catalysts for electrochemical N2 reduction toward NH3 under ambient conditions. ACS Catal. 2019, 9, 5245-5267. doi: 10.1021/acscatal.9b00994
Fryzuk, M. D.; Johnson, S. A. The continuing story of dinitrogen activation. Coord. Chem. Rev. 2000, 200, 379-409.
Lv, Z. J.; Wei, J. N.; Zhang, W. X.; Chen, P.; Deng, D. H.; Shi, Z. J.; Xi, Z. F. Direct transformation of dinitrogen: synthesis of N-containing organic compounds via N-C bond formation. Natl. Sci. Rev. 2020, 7, 1564-1583. doi: 10.1093/nsr/nwaa142
Honkala, K.; Hellman, A.; Remediakis, IN.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C. H.; Norskov, J. K. Ammonia synthesis from first-principles calculations. Science 2005, 307, 555-558. doi: 10.1126/science.1106435
Logadottir, A.; Nørskov, J. K. Ammonia synthesis over a Ru (0001) surface studied by density functional calculations. J. Catal. 2003, 220, 273-279. doi: 10.1016/S0021-9517(03)00156-8
Hoffman, B. M.; Lukoyanov, D.; Yang, Z. Y.; Dean, D. R.; Seefeldt, L. C. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev. 2014, 114, 4041-4062. doi: 10.1021/cr400641x
Burford, R. J.; Fryzuk, M. D. Examining the relationship between coordination mode and reactivity of dinitrogen. Nat. Rev. Chem. 2017, 1, 0026. doi: 10.1038/s41570-017-0026
Musaev, D. G. Theoretical prediction of a new dinitrogen reduction process: utilization of four dihydrogen molecules and a Zr2Pt2 cluster. J. Phys. Chem. B 2004, 108, 10012-10018. doi: 10.1021/jp0482767
Yamabe, T.; Hori, K.; Minato, T.; Fukui, K. Theoretical study on the bonding nature of transition-metal complexes of molecular nitrogen. Inorg. Chem. 1980, 19, 2154-2159. doi: 10.1021/ic50209a063
Holland, P. L. Metal-dioxygen and metal-dinitrogen complexes: where are the electrons? Dalton Trans. 2010, 39, 5415-5425. doi: 10.1039/c001397h
Henderson, R. Activation of dinitrogen at binuclear sites. Transition Met. Chem. 1990, 15, 330-336. doi: 10.1007/BF01061944
Crossland, J. L.; Tyler, D. R. Iron-dinitrogen coordination chemistry: dinitrogen activation and reactivity. Coord. Chem. Rev. 2010, 254, 1883-1894. doi: 10.1016/j.ccr.2010.01.005
Pan, F. Can structural chemistry point the way: exploring the relevance between structure and properties. Chin. J. Struct. Chem. 2020, 39, 7.
Zhang, M. Y.; Zhang, Y. Y.; Zhang, H. X.; Wang, K.; Wang, Y. Q.; Zeng, Y. F.; Liu, G. Y. Synthesis, crystal structure and catalytic activity palladium (Ⅱ) complexes containing bulky azole ligands. Chin. J. Struct. Chem. 2020, 39, 1669-1674.
Chirik, P. J. Dinitrogen functionalization with bis(cyclopentadienyl) complexes of zirconium and hafnium. Dalton Trans. 2007, 16-25.
Pfirrmann, S.; Limberg, C.; Herwig, C.; Stößer, R.; Ziemer, B. A dinuclear nickel (Ⅰ) dinitrogen complex and its reduction in single-electron steps. Angew. Chem. Int. Ed. 2009, 48, 3357-3361. doi: 10.1002/anie.200805862
Betley, T. A.; Peters, J. C. Dinitrogen chemistry from trigonally coordinated iron and cobalt platforms. J. Am. Chem. Soc. 2003, 125, 10782-10783. doi: 10.1021/ja036687f
Holland, P. L. Electronic structure and reactivity of three-coordinate iron complexes. Acc. Chem. Res. 2008, 41, 905-914. doi: 10.1021/ar700267b
Lu, J. B.; Ma, X. L.; Wang, J. Q.; Jiang, Y. F.; Li, Y.; Hu, H. S.; Xiao, H.; Li, J. The df-d dative bonding in a uranium-cobalt heterobimetallic complex for efficient nitrogen fixation. Inorg. Chem. 2019, 58, 7433-7439. doi: 10.1021/acs.inorgchem.9b00598
Ferreira, R. B.; Murray, L. J. Cyclophanes as platforms for reactive multimetallic complexes. Acc. Chem. Res. 2019, 52, 447-455. doi: 10.1021/acs.accounts.8b00559
Eaton, M. C.; Catalano, V. J.; Shearer, J.; Murray, L. J. Dinitrogen insertion and cleavage by a metal-metal bonded tricobalt (Ⅰ) cluster. J. Am. Chem. Soc. 2021, 143, 5649-5653. doi: 10.1021/jacs.1c01840
Allen, A. D.; Senoff, C. V. Nitrogenopentammineruthenium (Ⅱ) complexes. Chem. Commun. (London) 1965, 621-622.
Harrison, D. F.; Weissberger, E.; Taube, H. Binuclear ion containing nitrogen as a bridging group. Science 1968, 159, 320-322. doi: 10.1126/science.159.3812.320
Pun, D.; Lobkovsky, E.; Chirik, P. J. Indenyl zirconium dinitrogen chemistry: N2 coordination to an isolated zirconium sandwich and synthesis of side-on, end-on dinitrogen compounds. J. Am. Chem. Soc. 2008, 130, 6047-6054. doi: 10.1021/ja801021w
Fryzuk, M. D. Side-on end-on bound dinitrogen: an activated bonding mode that facilitates functionalizing molecular nitrogen. Acc. Chem. Res. 2009, 42, 127-133. doi: 10.1021/ar800061g
Chalkley, M. J.; Drover, M. W.; Peters, J. C. Catalytic N2-to-NH3 (or-N2H4) conversion by well-defined molecular coordination complexes. Chem. Rev. 2020, 120, 5582-5636. doi: 10.1021/acs.chemrev.9b00638
Yandulov, D. V.; Schrock, R. R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301, 76-78. doi: 10.1126/science.1085326
Arashiba, K.; Miyake, Y.; Nishibayashi, Y. A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat. Chem. 2011, 3, 120-125. doi: 10.1038/nchem.906
Anderson, J. S.; Rittle, J.; Peters, J. C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84-87. doi: 10.1038/nature12435
Takahashi, T.; Mizobe, Y.; Sato, M.; Uchida, Y.; Hidai, M. Protonation reactions of molybdenum and tungsten dinitrogen complexes with halogen acids. Hydride hydrazido (2-) and diazenido complexes as intermediate stages of reduction. J. Am. Chem. Soc. 1980, 102, 7461-7467. doi: 10.1021/ja00545a011
Oshita, H.; Mizobe, Y.; Hidai, M. Preparation and properties of molybdenum and tungsten dinitrogen complexes: XLI*. Silylation and germylation of a coordinated dinitrogen in cis-[M(N2)2(PMe2Ph)4] (M = Mo, W) using R3ECl/NaI and R3ECl/Na mixtures (E = Si, Ge). X-ray structure of trans-[WI(NNGePh3)(PMe2Ph)4]·C6H6. J. Organomet. Chem. 1993, 456, 213-220. doi: 10.1016/0022-328X(93)80428-E
Hidai, M.; Mizobe, Y. Recent advances in the chemistry of dinitrogen complexes. Chem. Rev. 1995, 95, 1115-1133. doi: 10.1021/cr00036a008
Tanabe, Y.; Nishibayashi, Y. Catalytic dinitrogen fixation to form ammonia at ambient reaction conditions using transition metal-dinitrogen complexes. Chem. Rec. 2016, 16, 1549-1577. doi: 10.1002/tcr.201600025
Lee, Y.; Mankad, N. P.; Peters, J. C. Triggering N2 uptake via redox-induced expulsion of coordinated NH3 and N2 silylation at trigonal bipyramidal iron. Nat. Chem. 2010, 2, 558-565. doi: 10.1038/nchem.660
Moret, M. -E.; Peters, J. C. N2 functionalization at iron metallaboratranes. J. Am. Chem. Soc. 2011, 133, 18118-18121. doi: 10.1021/ja208675p
Suess, D. L.; Peters, J. C. H-H and Si-H bond addition to Fe≡NNR2 intermediates derived from N2. J. Am. Chem. Soc. 2013, 135, 4938-4941. doi: 10.1021/ja400836u
Ung, G.; Peters, J. C. Low-temperature N2 binding to two-coordinate L2Fe0 enables reductive trapping of L2FeN2- and NH3 generation. Angew. Chem. Int. Ed. 2015, 54, 532-535.
Higuchi, J.; Kuriyama, S.; Eizawa, A.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Preparation and reactivity of iron complexes bearing anionic carbazole-based PNP-type pincer ligands toward catalytic nitrogen fixation. Dalton Trans. 2018, 47, 1117-1121. doi: 10.1039/C7DT04327A
Bezdek, M. J.; Guo, S.; Chirik, P. J. Terpyridine molybdenum dinitrogen chemistry: synthesis of dinitrogen complexes that vary by five oxidation states. Inorg. Chem. 2016, 55, 3117-3127. doi: 10.1021/acs.inorgchem.6b00053
Klopsch, I.; Finger, M.; Wurtele, C.; Milde, B.; Werz, D. B.; Schneider, S. Dinitrogen splitting and functionalization in the coordination sphere of rhenium. J. Am. Chem. Soc. 2014, 136, 6881-6883. doi: 10.1021/ja502759d
Tanaka, H.; Nishibayashi, Y.; Yoshizawa, K. Interplay between theory and experiment for ammonia synthesis catalyzed by transition metal complexes. Acc. Chem. Res. 2016, 49, 987-995. doi: 10.1021/acs.accounts.6b00033
Fryzuk, M. D.; Kozak, C. M.; Bowdridge, M. R.; Patrick, B. O.; Rettig, S. J. Nitride formation by thermolysis of a kinetically stable niobium dinitrogen complex. J. Am. Chem. Soc. 2002, 124, 8389-8397. doi: 10.1021/ja025997f
Evans, W. J.; Chamberlain, L.; Ulibarri, T. A.; Ziller, J. W. Reactivity of trimethylaluminum with (C5Me5)2Sm(THF)2: synthesis, structure, and reactivity of the samarium methyl complexes (C5Me5)2Sm[(μ-Me)AlMe2-(μ-Me)]2Sm(C5Me5)2 and (C5Me5)2SmMe(THF). J. Am. Chem. Soc. 1988, 110, 6423-6432. doi: 10.1021/ja00227a023
MacLachlan, E. A.; Fryzuk, M. D. Synthesis and reactivity of side-on-bound dinitrogen metal complexes. Organometallics 2006, 25, 1530-1543. doi: 10.1021/om051055i
Ma, X.; Tang, Y.; Lei, M. Bent and planar structures of μ-η2: η2-N2 dinuclear early transition metal complexes. Dalton Trans. 2014, 43, 11658-11666. doi: 10.1039/C4DT00646A
Ding, K. Y.; Pierpont, A. W.; Brennessel, W. W.; Lukat-Rodgers, G.; Rodgers, K. R.; Cundari, T. R.; Bill, E.; Holland, P. L. Cobalt-dinitrogen complexes with weakened N-N bonds. J. Am. Chem. Soc. 2009, 131, 9471-9472. doi: 10.1021/ja808783u
Fryzuk, M. D.; Love, J. B.; Rettig, S. J.; Young, V. G. Transformation of coordinated dinitrogen by reaction with dihydrogen and primary silanes. Science 1997, 275, 1445-1447. doi: 10.1126/science.275.5305.1445
Falcone, M.; Chatelain, L.; Scopelliti, R.; Živković, I.; Mazzanti, M. Nitrogen reduction and functionalization by a multimetallic uranium nitride complex. Nature 2017, 547, 332-335. doi: 10.1038/nature23279
Fryzuk, M. D.; Johnson, S. A.; Rettig, S. J. New mode of coordination for the dinitrogen ligand: a dinuclear tantalum complex with a bridging N2 unit that is both side-on and end-on. J. Am. Chem. Soc. 1998, 120, 11024-11025. doi: 10.1021/ja982377z
Fryzuk, M. D.; Johnson, S. A.; Patrick, B. O.; Albinati, A.; Mason, S. A.; Koetzle, T. F. New mode of coordination for the dinitrogen ligand: formation, bonding, and reactivity of a tantalum complex with a bridging N2 unit that is both side-on and end-on. J. Am. Chem. Soc. 2001, 123, 3960-3973. doi: 10.1021/ja0041371
Figg, T. M.; Holland, P. L.; Cundari, T. R. Cooperativity between low-valent iron and potassium promoters in dinitrogen fixation. Inorg. Chem. 2012, 51, 7546-7550. doi: 10.1021/ic300150u
MacLeod, K. C.; Vinyard, D. J.; Holland, P. L. A multi-iron system capable of rapid N2 formation and N2 cleavage. J. Am. Chem. Soc. 2014, 136, 10226-10229. doi: 10.1021/ja505193z
Singh, D.; Buratto, W. R.; Torres, J. F.; Murray, L. J. Activation of dinitrogen by polynuclear metal complexes. Chem. Rev. 2020, 120, 5517-5581. doi: 10.1021/acs.chemrev.0c00042
Smith, J. M.; Sadique, A. R.; Cundari, T. R.; Rodgers, K. R.; Lukat-Rodgers, G.; Lachicotte, R. J.; Flaschenriem, C. J.; Vela, J.; Holland, P. L. Studies of low-coordinate iron dinitrogen complexes. J. Am. Chem. Soc. 2006, 128, 756-769. doi: 10.1021/ja052707x
Chiang, K. P.; Bellows, S. M.; Brennessel, W. W.; Holland, P. L. Multimetallic cooperativity in activation of dinitrogen at iron-potassium sites. Chem. Sci. 2014, 5, 267-274. doi: 10.1039/C3SC52487F
McWilliams, S. F.; Holland, P. L. Dinitrogen binding and cleavage by multinuclear iron complexes. Acc. Chem. Res. 2015, 48, 2059-2065. doi: 10.1021/acs.accounts.5b00213
Reiners, M.; Baabe, D.; Munster, K.; Zaretzke, M. K.; Freytag, M.; Jones, P. G.; Coppel, Y.; Bontemps, S.; del Rosal, I.; Maron, L.; Walter, M. D. NH3 formation from N2 and H2 mediated by molecular tri-iron complexes. Nat. Chem. 2020, 12, 740-746. doi: 10.1038/s41557-020-0483-7
Xin, X.; Douair, I.; Zhao, Y.; Wang, S.; Maron, L.; Zhu, C. Dinitrogen cleavage by a heterometallic cluster featuring multiple uranium-rhodium bonds. J. Am. Chem. Soc. 2020, 142, 15004-15011. doi: 10.1021/jacs.0c05788
Jori, N.; Barluzzi, L.; Douair, I.; Maron, L.; Fadaei-Tirani, F.; Zivkovic, I.; Mazzanti, M. Stepwise reduction of dinitrogen by a uranium-potassium complex yielding a U(Ⅵ)/U(Ⅳ) tetranitride cluster. J. Am. Chem. Soc. 2021, 143, 11225-11234. doi: 10.1021/jacs.1c05389
Forrest, S. J.; Schluschaß, B.; Yuzik-Klimova, E. Y.; Schneider, S. Nitrogen fixation via splitting into nitrido complexes. Chem. Rev. 2021, 121, 6522-6587. doi: 10.1021/acs.chemrev.0c00958
Cui, C. N.; Zhang, H. Y.; Luo, Z. X.; Pan, F. Preparation and reaction of naked metal clusters for catalysis and genetic materials. Chin. J. Struct. Chem. 2020, 39, 989-998.
Han, Y.; Jiang, Y.; Yang, J. J.; Lin, S. C.; Tang, Z. C.; Zheng, L. S. Tuning solvent composition to enhance the stability of metal clusters in mass spectrometry. Chin. J. Struct. Chem. 2022, 41, 2204034-2204039.
Kuganathan, N.; Green, J. C.; Himmel, H. -J. Dinitrogen fixation and activation by Ti and Zr atoms, clusters and complexes. New J. Chem. 2006, 30, 1253-1262. doi: 10.1039/b606328d
Zhao, Y.; Cui, J. T.; Wang, M.; Valdivielso, D. Y.; Fielicke, A.; Hu, L. R.; Cheng, X.; Liu, Q. Y.; Li, Z. Y.; He, S. G.; Ma, J. B. Dinitrogen fixation and reduction by Ta3N3H0, 1- cluster anions at room temperature: hydrogen-assisted enhancement of reactivity. J. Am. Chem. Soc. 2019, 141, 12592-12600. doi: 10.1021/jacs.9b03168
Li, Z. Y.; Mou, L. H.; Wei, G. P.; Ren, Y.; Zhang, M. Q.; Liu, Q. Y.; He, S. G. C-N coupling in N2 fixation by the ditantalum carbide cluster anions Ta2C4-. Inorg. Chem. 2019, 58, 4701-4705. doi: 10.1021/acs.inorgchem.8b03502
Geng, C.; Li, J.; Weiske, T.; Schwarz, H. Complete cleavage of the N≡N triple bond by Ta2N+ via degenerate ligand exchange at ambient temperature: a perfect catalytic cycle. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 21416-21420. doi: 10.1073/pnas.1913664116
Li, Z. Y.; Li, Y.; Mou, L. H.; Chen, J. J.; Liu, Q. Y.; He, S. G.; Chen, H. A facile N≡N bond cleavage by the trinuclear metal center in vanadium carbide cluster anions V3C4-. J. Am. Chem. Soc. 2020, 142, 10747-10754. doi: 10.1021/jacs.0c02021
Wang, X. Y.; Peng, X. B.; Zhang, Y. F.; Ni, J.; Au, C. T.; Jiang, L. L. Efficient ammonia synthesis over a core-shell Ru/CeO2 catalyst with a tunable CeO2 size: DFT calculations and XAS spectroscopy studies. Inorg. Chem. Front. 2019, 6, 396-406. doi: 10.1039/C8QI01244J
Himmel, H. J.; Hübner, O.; Klopper, W.; Manceron, L. Cleavage of the N2 triple bond by the Ti dimer: a route to molecular materials for dinitrogen activation? Angew. Chem. Int. Ed. 2006, 45, 2799-2802. doi: 10.1002/anie.200503709
Zhou, M.; Jin, X.; Gong, Y.; Li, J. Remarkable dinitrogen activation and cleavage by the Gd dimer: from dinitrogen complexes to ring and cage nitrides. Angew. Chem. Int. Ed. 2007, 46, 2911-2914. doi: 10.1002/anie.200605218
Gong, Y.; Zhao, Y.; Zhou, M. Formation and characterization of the tetranuclear scandium nitride: Sc4N4. J. Phys. Chem. A. 2007, 111, 6204-6207. doi: 10.1021/jp070816n
Jiang, G. D.; Li, Z. Y.; Mou, L. H.; He, S. G. Dual iron sites in activation of N2 by iron-sulfur cluster anions Fe5S2- and Fe5S3-. J. Phys. Chem. Lett. 2021, 12, 9269-9274. doi: 10.1021/acs.jpclett.1c02683
Mou, L. H.; Li, Y.; Li, Z. Y.; Liu, Q. Y.; Ren, Y.; Chen, H.; He, S. G. Dinitrogen activation and functionalization by heteronuclear metal cluster anions FeV2C2- at room temperature. J. Phys. Chem. Lett. 2020, 11, 9990-9994. doi: 10.1021/acs.jpclett.0c02921
Ma, X. L.; Liu, J. C.; Xiao, H.; Li, J. Surface single-cluster catalyst for N2-to-NH3 thermal conversion. J. Am. Chem. Soc. 2018, 140, 46-49. doi: 10.1021/jacs.7b10354
Liu, J. C.; Ma, X. L.; Li, Y.; Wang, Y. G.; Xiao, H.; Li, J. Heterogeneous Fe3 single-cluster catalyst for ammonia synthesis via an associative mechanism. Nat. Commun. 2018, 9, 1610. doi: 10.1038/s41467-018-03795-8
Cherkasov, N.; Ibhadon, A.; Fitzpatrick, P. A review of the existing and alternative methods for greener nitrogen fixation. Chem. Eng. Process. 2015, 90, 24-33. doi: 10.1016/j.cep.2015.02.004
Zhang, X.; Chen, A.; Zhang, Z.; Zhou, Z. Double-atom catalysts: transition metal dimer-anchored C2N monolayers as N2 fixation electrocatalysts. J. Mater. Chem. A 2018, 6, 18599-18604. doi: 10.1039/C8TA07683A
Chen, Z. W.; Yan, J. M.; Jiang, Q. Single or double: which is the altar of atomic catalysts for nitrogen reduction reaction? Small Methods 2019, 3, 1800291. doi: 10.1002/smtd.201800291
Han, B.; Meng, H.; Li, F.; Zhao, J. Fe3 cluster anchored on the C2N monolayer for efficient electrochemical nitrogen fixation. Catalysts 2020, 10, 974. doi: 10.3390/catal10090974
Ma, D.; Zeng, Z.; Liu, L.; Huang, X.; Jia, Y. Computational evaluation of electrocatalytic nitrogen reduction on TM single-, double-, and triple-atom catalysts (TM = Mn, Fe, Co, Ni) based on graphdiyne monolayers. J. Phys. Chem. C 2019, 123, 19066-19076. doi: 10.1021/acs.jpcc.9b05250
Li, M.; Cui, Y.; Zhang, X.; Luo, Y.; Dai, Y.; Huang, Y. Screening a suitable Mo form supported on graphdiyne for effectively electrocatalytic N2 reduction reaction: from atomic catalyst to cluster catalyst. J. Phys. Chem. Lett. 2020, 11, 8128-8137. doi: 10.1021/acs.jpclett.0c02354
Chen, Z. W.; Chen, L. X.; Jiang, M.; Chen, D.; Wang, Z. L.; Yao, X.; Singh, C. V.; Jiang, Q. A triple atom catalyst with ultrahigh loading potential for nitrogen electrochemical reduction. J. Mater. Chem. A 2020, 8, 15086-15093. doi: 10.1039/D0TA04919K
Zheng, G.; Li, L.; Tian, Z.; Zhang, X.; Chen, L. Heterogeneous single-cluster catalysts (Mn3, Fe3, Co3, and Mo3) supported on nitrogen-doped graphene for robust electrochemical nitrogen reduction. J. Energy Chem. 2021, 54, 612-619. doi: 10.1016/j.jechem.2020.06.048
Cui, C. N.; Zhang, H. C.; Luo, Z. X. Nitrogen reduction reaction on small iron clusters supported by N-doped graphene: a theoretical study of the atomically precise active-site mechanism. Nano Res. 2020, 13, 2280-2288. doi: 10.1007/s12274-020-2847-0
Zhang, H. C.; Cui, C. N.; Luo, Z. X. MoS2-supported Fe2 clusters catalyzing nitrogen reduction reaction to produce ammonia. J. Phys. Chem. C 2020, 124, 6260-6266. doi: 10.1021/acs.jpcc.0c00486
Yao, C. H.; Guo, N.; Xi, S. B.; Xu, C. Q.; Liu, W.; Zhao, X. X.; Li, J.; Fang, H. Y.; Su, J.; Chen, Z. X. Atomically-precise dopant-controlled single cluster catalysis for electrochemical nitrogen reduction. Nat. Commun. 2020, 11, 4389. doi: 10.1038/s41467-020-18080-w
Ma, X. L.; Yang, Y.; Xu, L. M.; Xiao, H.; Yao, W. Z.; Li, J. Theoretical investigation on hydrogenation of dinitrogen triggered by singly dispersed bimetallic sites. J. Mater. Chem. A 2022, 10, 6146-6152. doi: 10.1039/D1TA08350C
Luo, Y.; Li, M.; Dai, Y.; Zhao, R.; Jiang, F.; Wang, S.; Huang, Y. Transition metal-modified Co4 clusters supported on graphdiyne as an effective nitrogen reduction reaction electrocatalyst. Inorg. Chem. 2021, 60, 18251-18259. doi: 10.1021/acs.inorgchem.1c02880
Wang, Q.; Pan, J.; Guo, J.; Hansen, H. A.; Xie, H.; Jiang, L.; Hua, L.; Li, H.; Guan, Y.; Wang, P. Ternary ruthenium complex hydrides for ammonia synthesis via the associative mechanism. Nat. Catal. 2021, 4, 959-967. doi: 10.1038/s41929-021-00698-8
Liu, C.; Wang, Q.; Guo, J.; Vegge, T.; Chen, P.; Hansen, H. A. Formation of a complex active center by Ba2RuH6 for nondissociative dinitrogen activation and ammonia formation. ACS Catal. 2022, 12, 4194-4202. doi: 10.1021/acscatal.2c00180
Wang, X. Y.; Li, L. L.; Fang, Z. P.; Zhang, Y. F.; Ni, J.; Lin, B. Y.; Zheng, L. R.; Au, C. -T.; Jiang, L. L. Atomically dispersed Ru catalyst for low-temperature nitrogen activation to ammonia via an associative mechanism. ACS Catal. 2020, 10, 9504-9514. doi: 10.1021/acscatal.0c00549
Wang, X. Y.; Peng, X. B.; Chen, W.; Liu, G. Y.; Zheng, A. M.; Zheng, L. R.; Ni, J.; Au, C. T.; Jiang, L. L. Insight into dynamic and steady-state active sites for nitrogen activation to ammonia by cobalt-based catalyst. Nat. Commun. 2020, 11, 653. doi: 10.1038/s41467-020-14287-z
Hu, H. S.; Xu, X.; Xu, C.; Li. J. Recent progresses in experimental and theoretical studies of actinide clusters. Chin. J. Struct. Chem. 2020, 39, 1201-1212.
Liu, J. C.; Xiao, H.; Zhao, X. K.; Zhang, N. N.; Liu, Y.; Xing, D. H.; Yu, X. H.; Hu, H. S.; Li, J. Computational prediction of graphdiyne-supported three-atom single-cluster catalysts. CCS Chem. 2022, DOI: 10.31635/ccschem.022.202201796.

Figure 2 (a) Schematic illustration of the binding modes of N2 on mono-nuclear or multi-nuclear complexes or clusters. (b) Schematic binding modes of N2 in trinuclear complexes or clusters. Note that no distinction was made between single, double or triple bonds. The nomenclatures used in the text are the abbreviation for the coordination mode of "Terminal" (T), "Bridging" (B), end-on (E) and side-on (S). For instance, "BESS" stands for the bridging end-on/side-on/side-on binding mode. The pink balls stand for the dinitrogen active site in the metal complexes or clusters.

Figure 3 N2 activation and functionalization mechanism based on different N2 binding modes. Reproduced with permission from Ref.[11] Copyright 2017, Nature Publishing Group.

Figure 4 N2 adsorption configuration on different surface single-cluster catalysts (SCCs) for ammonia synthesis. Reproduced with permission from Ref. [78-91] Copyright 2018, American Chemical Society; Copyright 2018, Nature Publishing Group; Copyright 2015, Elsevier; Copyright 2018, Royal Society of Chemistry; Copyright 2019, WILEY-VCH; Copyright 2020, MDPI; Copyright 2019, America Chemical Society; Copyright 2020, America Chemical Society; Copyright 2020, Royal Society of Chemistry; Copyright 2021, Elsevier; Copyright 2020, Tsinghua University Press; Copyright 2020, America Chemical Society; Copyright 2020, Nature Publishing Group and Copyright 2022, Royal Society of Chemistry.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
