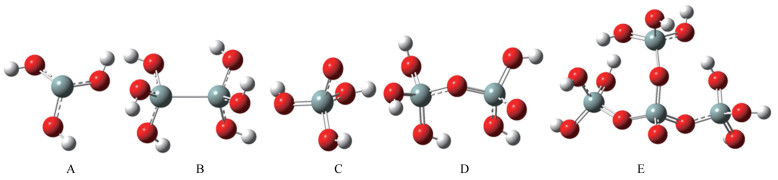
图 1.
几种典型的SiO2分子簇模型
Figure 1.
Several typical SiO2 cluster models

随着中国经济的稳步增长以及工业化、城镇化进程的加快,社会发展对能源的需求不断增加。煤炭作为中国能源的主要来源之一,其支柱地位还将持续相当长的一段时间[1]。煤炭是一种极其复杂的物质,除含有C、H、O、N和S等主量元素外,还含有Al、Si、Fe、Na、K和Ca等次量元素和Hg、As、Se、Pb和Cd等对环境危害颇大的痕量元素。针对其燃烧过程中痕量元素排放的研究,已成为燃烧污染领域一个新的前沿课题。砷是当前环境中对人体危害最大的痕量元素之一,具有致癌作用,由于砷具有挥发性,因此, 煤燃烧过程中约1/3的砷会直接挥发并排入大气中,另外砷极易富集于微小颗粒上,可以长距离传输造成大范围污染[2]。尽管砷在煤中的含量较低,但是由于燃煤过程中煤耗量巨大,煤中砷的长期排放和积累对人体和环境产生的危害不容忽视。
近年来,一些学者对煤燃烧过程中砷的形态分布、排放和迁移规律进行了研究。王泉海等[3]研究发现,在较高温度的氧化性气氛中,砷及其化合物主要以气态形式存在, 在900 K温度以上,砷主要以气态AsO形态存在, 在800 K左右的温度区间内,砷以气态AsO、固态As2O5和As4O6形态存在, 温度低于800 K时,砷以固态As2O5形式存在;Ding等[4]发现在锅炉燃烧高温区(1300-1400 ℃),伴随着煤中有机质的燃烧和矿物质发生的破碎、分解、熔融等反应,各种赋存形式的砷将主要以气相氧化砷(AsO)形式存在, 残留于底渣和炉渣中的量很少, 砷大部分(90%)均富集在灰中,其中, 飞灰中的主量无机成分被认为是砷吸附的主要成因。同时董静兰等[5]对于煤与生物质掺烧时污染物砷排放的研究表明, 掺烧生物质后砷的释放受到一定的抑制,生物质掺烧比例越大,灰中砷元素的相对富集系数越大。
对于飞灰中痕量元素的富集特性,研究者们尝试从微观层面予以解释:高正阳等[6, 7]分别基于密度泛函理论对炭基模型和MnO2改性活性炭模型吸附气态汞的机理进行了研究,郭欣等[8]对CaO表面对Hg和HgCl2的吸附机理进行了DFT研究。其中,炭基吸附剂主要是指未燃尽炭,而飞灰中的未燃尽炭含量很少(2%-12%)[9],飞灰中的CaO含量也较少(20%-40%)[10]。然而无论是燃煤飞灰还是生物质灰中都存在大量的SiO2,生物质稻壳灰中的SiO2含量更是高达86.5%,统计表明,中国火电厂粉煤灰的主要氧化物组成分别为:SiO2、CaO、K2O等,其含量顺序也大致遵循上述排列[11]。
飞灰中的SiO2主要以无定型形式存在[12],在这些无定型SiO2表面,既存在规则位置,也存在活性缺陷位,研究发现,规则位置活性很低,基本不与分子发生反应,而缺陷位置则具有较大的反应活性。前人的研究也得到同样的结论:Nuria等[13]在研究Cu、Pd和Cs在SiO2表面规则位置和缺陷位置的吸附时,发现三种原子在规则位置基本不反应,而在缺陷位置均能发生吸附反应,相比之下,缺陷部位如非桥接氧位≡Si-O·和Si悬挂键位≡Si·,活性较大,经验证其确实为成核发生的中心,确认Cu与表面规则位置的结合很弱并且仅在缺陷部位黏附这一事实;Ferullo等[14]在研究Cu、Ag和Au颗粒在SiO2上的吸附时,也发现这三种金属,无论是单体还是聚合体,基本不与表面规则位置反应,而均能在活泼的缺陷位沉积。
为了探究砷在飞灰中富集的原因,本研究采用密度泛函理论研究了砷的主要氧化物AsO在有缺陷位的无定型SiO2表面的吸附机理,通过对多种吸附构型的优化及能量计算等,得到AsO在飞灰中主要成分SiO2表面的吸附规律,为电厂飞灰的资源化利用、开发廉价高效的砷吸附剂提供一定的参考。
本研究选取的典型无定型SiO2分子的缺陷位簇模型是根据Nuria等[13]和Ferullo等[14]提出的模型建立的,具体见图 1。这些缺陷模型已经被证明既能反映催化剂的表面特征,又能较方便地实现计算和保证足够的量化计算精度。簇模型本质上是局部性的,不仅可以应用于块状晶体,而且可以很好地应用于无定形材料表面和复杂的界面[15]。所有簇模型均采用氢封闭,该方法饱和了簇模型中多余的悬空键,较好地考虑了簇与环境的短程作用,因而更适应于键方向性明确的共价型氧化物。对于SiO2共价固体,Si-O之间轨道重叠成键,键电子定域于原子之间,切取SiO2簇模型势必切断Si-O键,则用氢封闭消除悬空键成为必须。
模型主要包括:一为Si单独占据sp3的悬空键位:≡Si·,用(HO)3Si·模型表示,如图 1中A所示;二为中性氧空位:≡Si-Si≡,用(HO)3Si-Si(OH)3表示,如图 1中B所示;三为非桥接氧中心位:≡Si-O·,用(HO)3Si-O·和(HO)3Si-O-(HO)2Si-O·以及Si4O4(OH)9模型表示,分别如图 1中的C、D、E所示(红色小球为O,灰色小球为Si,白色小球为H)。
结构优化和频率计算采用密度泛函理论中的B3LYP方法[16]和6-311G(d)基组[17],能量计算采用密度泛函理论中的B3LYP方法和def2-TZVP[18]。结构优化、频率和能量计算通过Gaussian 09程序完成[19]。吸附构型的Atoms in Molecule (AIM)理论分析及构型中As、Si、O原子的Mulliken电荷计算,以及分析相互作用时所绘制的LOL填色图均通过波函数分析程序Multiwfn[20]完成,吸附构型的拓扑分析图通过Multiwfn分析程序结合VMD[21]程序绘制。
AsO在SiO2模型表面上的吸附能通过公式(1)计算:
|
${E_{{\rm{ads}}}} = {E_{{\rm{AB}}}}{\rm{ - }}{E_{\rm{A}}} - {E_{\rm{B}}} $ |
(1) |
式中,Eads为吸附能,EAB为复合物的能量,EA为单体A的能量,EB为单体B的能量。
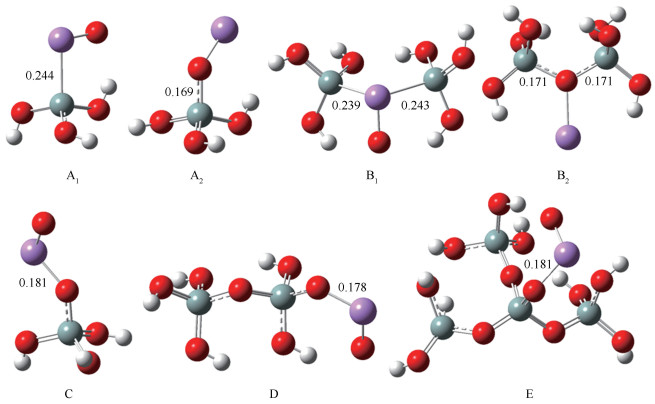
AsO在SiO2单体表面不同位置的吸附构型见图 2(紫色原子为As,红色原子为O,灰色原子为Si),图 2中标出了相互作用的两原子间的键长值(单位:nm)。同时考察了吸附构型的吸附能与吸附热,吸附能可以衡量吸附强弱,而吸附热更加对应着实际的可测量量,吸附能和吸附热数值越大,吸附能力越强。不同吸附构型的吸附能与吸附热数值分别见表 1、表 2。
 下载:
导出CSV
下载:
导出CSV
| Configuration | Eads /(kJ·mol-1) | Configuration | Eads /(kJ·mol-1) | ||
| SiO2-AsO | A1 | -214.14 | SiO2-AsO | C | -386.76 |
| A2 | -248.49 | D | -408.62 | ||
| B1 | -83.04 | E | -427.50 | ||
| B2 | -210.77 | ||||
下载:
导出CSV
| Configuration | Hads /(kJ·mol-1) | Configuration | Hads /(kJ·mol-1) | ||
| SiO2-AsO | A1 | 225.08 | SiO2-AsO | C | 376.52 |
| A2 | 250.65 | D | 404.94 | ||
| B1 | 84.00 | E | 432.04 | ||
| B2 | 217.07 | ||||
由表 1可知,这七种吸附构型的吸附能均为负值,绝对值均大于50 kJ/mol,说明SiO2单体表面吸附AsO形成的复合物为稳定构型,且为化学吸附。B构型中SiO2中的缺陷位为中性氧空位,当中性氧空位与AsO中的As相互作用时,吸附能较小,仅为83.04 kJ/mol;而当中性氧空位与AsO中的O相互作用时,吸附能较大,为210.77 kJ/mol,说明AsO中的O原子更易与SiO2中的中性氧空位反应。A1、A2、C、D、E吸附构型的吸附能很大, 范围在214.14 -427.50 kJ/mol,为较强的化学吸附。说明Si单独占据sp3的悬空键位≡Si·和非桥接氧中心位≡Si-O·这两种缺陷位的活性很大,且非桥接氧中心位≡Si-O·比Si单独占据sp3的悬空键位≡Si·活性更大,一方面,前者吸附构型中的吸附能明显大于后者;另一方面,O的电负性要明显大于Si,其中,Si电负性为1.90,而O电负性为3.44,元素的电负性越大,其原子在化合物中吸引电子的能力越强,因此,活性更大。C、D和E均属于非桥接氧中心位≡Si-O·中的构型,三者的构型一致,区别就是分子簇的大小,C的吸附能为386.76 kJ/mol,D的吸附能为408.62 kJ/mol,E的吸附能为427.50 kJ/mol,说明同种构型下,分子簇越大,吸附能也越大。
由表 2可知,其规律基本与表 1一致,即同样得到:非桥接氧中心位≡Si-O·对应的C、D、E三种吸附构型的吸附热大,吸附强;其次是Si单独占据sp3的悬空键位≡Si·和中性氧空位对应的吸附构型。并且具有这三种缺陷位的SiO2与AsO之间均为强相互作用。
AIM理论认为,临界点中的键鞍点(bond critical point, BCP)表明两个原子之间存在相互作用,且这个点的性质(电子密度、能量密度、动能密度等)可以用来讨论成键特征[22]。键临界点处电子密度的拉普拉斯值可以用来初步判断相互作用的类型,正值和负值分别表示闭壳层和共价键相互作用。键临界点处电子密度ρ(r)值越大,说明该化学键的强度越大;反之越小。
Cremer等[23]提出,可以根据键临界点的势能密度(V(r))、拉格朗日动能密度(G(r))和总能量密度(H(r))来判断键的种类,对于共价相互作用,|V(r)| > G(r)并且H(r) < 0;对于闭壳层相互作用|V(r)| < G(r)并且H(r) > 0。
Espinosa等[24]提出了类似的判断准则,当|V(r)|/G(r)>2时,可以认定相互作用为共价键类型;当|V(r)|/G(r) < 1时可以认为是闭壳层相互作用。
七种SiO2分子簇与AsO吸附构型的拓扑图见图 3。
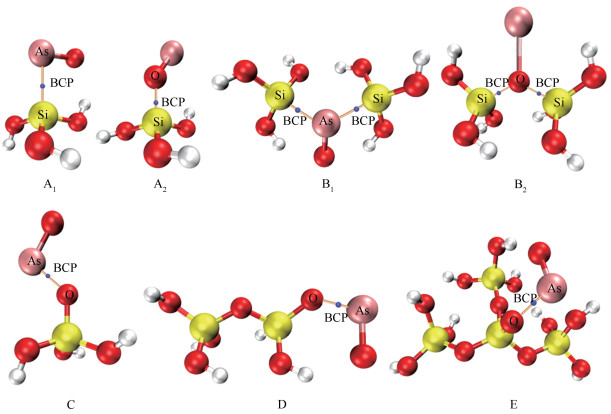
各吸附构型所有临界点的数量均满足Poincaré-Hopf关系,说明所有的CP都可能被找到。为便于分析SiO2-AsO之间的相互作用类型,只在拓扑图中显示两分子之间相互作用的原子间的键临界点(BCP)。由图 3的拓扑图可知,两分子之间相互作用的原子间均存在键临界点(BCP),说明这些原子间存在相互作用。
SiO2分子簇与AsO的吸附构型中Si-As键、Si-O键和O-As键的键临界点拓扑分析性质及主要原子的Mulliken电荷见表 3,下面针对每种构型,通过表 3的相应BCP点处的各类实空间函数值,对其相互作用类型进行分析。
下载:
导出CSV
| Configuration | Bond | ρ | ▽2ρ | G | V | H | |V|/G | Mulliken |
||
| As/O | △ | |||||||||
| SiO2-AsO | A1 | Si1-As8 | 0.242 | -0.013 | 0.085 | -0.154 | -0.204 | 2.12 | 0.150 | -0.322 |
| A2 | Si1-O9 | 0.249 | 0.342 | 0.089 | -0.183 | -0.344 | 2.15 | -0.831 | -0.359 | |
| B1 | Si8-As15 | 0.210 | 0.145 | 0.029 | -0.060 | -0.048 | 2.07 | 0.329 | -0.143 | |
| Si1-As15 | 0.211 | 0.134 | 0.029 | -0.061 | -0.047 | 2.10 | 0.329 | -0.143 | ||
| B2 | Si1-O16 | 0.242 | 0.385 | 0.074 | -0.153 | -0.203 | 2.08 | -0.793 | -0.321 | |
| Si8-O16 | 0.243 | 0.387 | 0.072 | -0.151 | -0.201 | 2.09 | -0.794 | -0.322 | ||
| C | O2-As9 | 0.290 | 0.359 | 0.104 | -0.229 | -0.565 | 2.20 | 0.947 | 0.475 | |
| D | O11-As15 | 0.321 | 0.406 | 0.135 | -0.348 | -0.733 | 2.58 | 0.954 | 0.482 | |
| E | O5-As27 | 0.323 | 0.343 | 0.152 | -0.419 | -0.665 | 2.76 | 1.043 | 0.571 | |
由表 2可知,在A1吸附构型中,Si-As键的键临界点处电子密度的拉普拉斯值▽2ρ小于零,表明SiO2-AsO之间为共价相互作用。此外,所有吸附构型的|V(r)|均大于G(r),且H(r)均小于零,根据Cremer提出的准则同样可以判断Si-As键、O-As键与Si-O键属于共价键,SiO2-AsO之间为共价相互作用。同时根据Espinoza提出的判断准则,表 2中所列出构型的|V(r)|/G(r)值均大于2.0,说明AsO通过共价键作用吸附在SiO2表面活性位上。而在除A1之外的吸附构型中,注意到两分子相互作用的原子之间所对应的键临界点处电子密度的拉普拉斯值▽2ρ却大于零,但利用BCP处电子密度拉普拉斯值的正负来判断相互作用类型有时会失效,对于其得出的矛盾结论,可以通过下述方法再次进行判断。
表 2中Mulliken电荷第一列数值为正值的为As原子在不同吸附构型中的Mulliken原子电荷,负值为O原子在不同吸附构型中的Mulliken原子电荷,第二列数值为As、O原子在吸附态AsO与孤立态AsO中的Mulliken原子电荷的差值。当差值为正值时,表明As、O原子失去电子;当差值为负值时,表明As、O原子得到电子。电荷转移量与相互作用能近似成正比关系,董兰等[25]在Be_n(n=1-6)团簇吸附H_2的密度泛函研究时,发现随着Be团簇原子数量增加,与H的s-p杂化程度增强,吸附位对应的电荷转移量越大,吸附越牢固;叶天旭等[26]在Co/MoS2催化剂加氢脱硫活性的量子化学研究中发现, 电荷转移量越大,对应的催化剂HDS活性越大,相互作用能越大。因此,As-Si、Si-O原子间以及As-O原子间的电荷转移量越多,形成的As-Si键以及As-O键的强度更高,对应的吸附能更大。并且发现A吸附构型和C吸附构型的分子簇大小相近,但是C构型的吸附能明显大于A,说明非桥接氧中心位≡Si-O·的活性要大于Si单独占据sp3的悬空键位≡Si·;而吸附构型D和B,其分子簇大小相近,但D的吸附能明显大于B,说明非桥接氧中心位:≡Si-O·的活性要大于中性氧空位:≡Si-Si≡,因此,在所考虑的三个主要缺陷位中,非桥接氧位≡Si-O·是最具反应活性的,其次是Si单独占据sp3的悬空键位≡Si·和中性氧空位≡Si-Si≡。该结论与Nuria等[13]的研究结果相同。
LOL函数是进行共价键分析的另外一种有力的工具,因为它揭示了发现电子对的可能性较高的分子空间区域[27]。LOL函数的取值一般为0-1。在0.5-1的较大数值区间内,表示含有键合和非键合的定域化电子的区域,而较小的值(<0.5)描述了电子为非定域化的,即为发散的。在电子密度主要受电子定域化程度支配的区域存在共价键,孤电子对或原子核壳层,因此,该区域具有较大的电子定域化程度,LOL的值也较高(>0.5)。
在这七种吸附构型中,在其LOL填色图中,可以明显看出两个分子中相互作用的原子Si-As、O-As、Si-O之间存在显著的电子定域性区域(图中两原子间的红色区域),说明AsO与SiO2之间为共价相互作用。
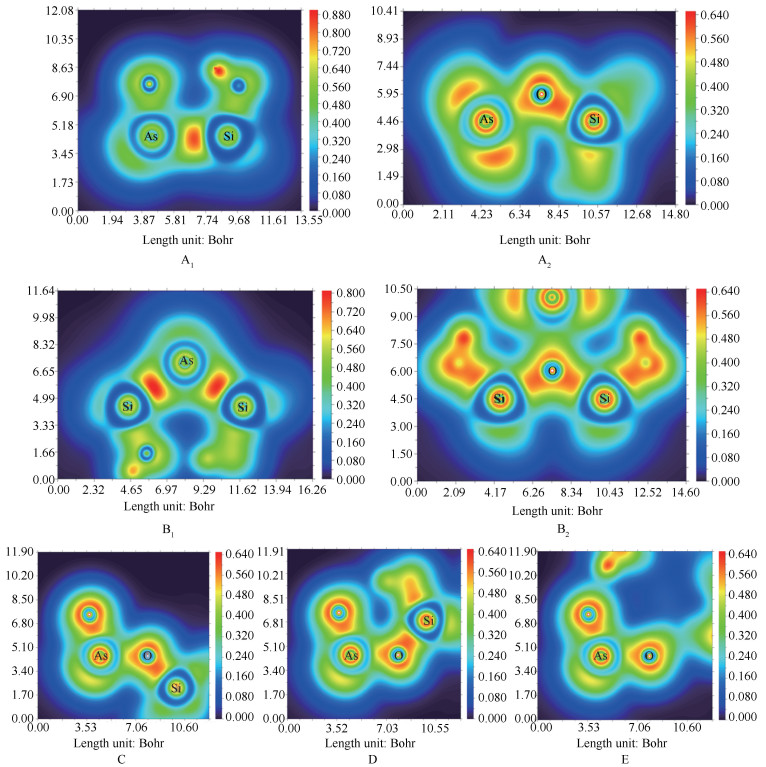
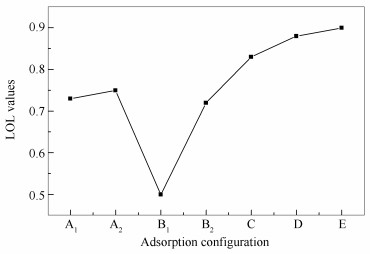
LOL填色图中只是定性地判断了AsO和SiO2分子之间的相互作用类型,下面通过使用Multiwfn对吸附构型的LOL函数做拓扑分析,并计算出图 4中用于判断相互作用类型所对应的BCP处的LOL值,对两分子间的相互作用类型进行定量分析。并将其结果绘制成曲线图,见图 5。由图 5可知,这七种吸附构型相应BCP处的LOL值均大于0.5,说明其相互作用类型为共价相互作用。此外,B1吸附构型的吸附能小于其他吸附构型的吸附能,从曲线上也可以看出B1构型相应区域的定域性程度相比其他几种构型低。
通过密度泛函理论对砷的典型氧化物AsO在无定型SiO2的几种典型的缺陷位模型表面的吸附机理进行了研究,通过对多种吸附构型进行结构优化、吸附能计算、AIM理论和Mulliken电荷分析,及绘制定域化轨道指示函数(LOL)填色图得到了AsO在SiO2表面的吸附规律。
存在缺陷位的SiO2分子模型表面,其活性较高,与AsO的吸附作用为典型的化学吸附,SiO2-AsO之间为共价相互作用,并且不同的缺陷位,其活性不同,非桥接氧中心位:≡Si-O·活性最大,其次是Si单独占据sp3的悬空键位:≡Si·>和中性氧空位:≡Si-Si≡。
通过AIM理论分析以及吸附构型的拓扑分析,发现SiO2-AsO之间参与相互作用的原子之间均存在键临界点,并且通过对各吸附构型中相互作用的两原子间对应的BCP处的实空间函数值分析,说明SiO2-AsO之间为共价相互作用。
通过Mulliken电荷分析,发现电荷转移量与相互作用能近似成正比关系。
通过LOL函数分析,发现SiO2-AsO之间相互作用的两原子间均存在较高的电子定域化区域,且该区域对应的LOL实空间函数值均大于0.5,亦说明SiO2-AsO之间为共价相互作用。
缺陷位SiO2被证明是一种有效的痕量元素吸附剂。在飞灰中含有大量的缺陷位SiO2,深入了解飞灰中的缺陷位SiO2吸附痕量元素砷的机理,为飞灰的资源化利用提供了理论基础,同时对于如何控制环境中的有害痕量气体排放提供了理论支持。
赵永椿, 马斯鸣, 杨建平. 燃煤电厂污染物超净排放的发展及现状[J]. 煤炭学报, 2015,40,(11): 2629-2640. ZHAO Yong-chun, MA Si-ming, YANG Jian-ping. Development and status quo of ultra-clean pollutant emissions from coal-fired power plants[J]. J China Coal Soc, 2015, 40(11): 2629-2640.
郭欣, 郑楚光, 陈丹. 300MW煤粉锅炉砷排放特征的实验研究[J]. 环境科学学报, 2006,27,(4): 631-634. GUO Xin, ZHENG Chu-guang, CHEN Dan. Experimental Study on arsenic emission characteristics of 300MW pulverized coal-fired boiler[J]. J Environ Sci-China, 2006, 27(4): 631-634.
王泉海, 邱建荣, 温存. 氧燃烧方式下痕量元素形态转化的试验和模拟研究[J]. 工程热物理学报, 2006,27,(s2): 199-202. WANG Quan-hai, QIU Jian-rong, WUN Cun. Experimental and simulation study on morphological transformation of trace elements under oxygen combustion[J]. J Eng Thermophys, 2006, 27(s2): 199-202.
DING Z H, ZHENG B S, ZHUANG M. Mode of occurrence of arsenic in high-As coals from endemic arsenicosis areas in southwestern Guizhou Province, China[J]. J Coal Sci Eng, 2007, 13(2): 194-198.
董静兰, 马凯. 富氧气氛下煤与生物质掺烧时砷的释放研究[J]. 动力工程学报, 2017,37,(3): 237-241. DONG Jing-lan, MA Kai. Study on arsenic release from coal-biomass combustion under oxygen-enriched atmosphere[J]. Chin J Power Eng, 2017, 37(3): 237-241.
高正阳, 吕少昆, 吉硕. 碳基表面吸附铅的机理[J]. 环境工程学报, 2016,10,(7): 3848-3852. GAO Zheng-yang, LÜ Shao-kun, JI Shuo. Mechanism of lead adsorption on carbon-based surfaces[J]. J Chin Environ Eng, 2016, 10(7): 3848-3852.
高正阳, 吕少昆. 二氧化锰改性活性炭吸附汞的机理研究[J]. 动力工程学报, 2015,35,(11): 923-928. doi: 10.3969/j.issn.1674-7607.2015.11.010GAO Zheng-yang, LÜ Shao-kun. Mechanism of adsorption of mercury by manganese dioxide modified activated carbon[J]. Chin J Power Eng, 2015, 35(11): 923-928. doi: 10.3969/j.issn.1674-7607.2015.11.010
郭欣, 郑楚光, 吕乃霞. 簇模型CaO(001)面上吸附汞与氯化汞的密度泛函理论研究[J]. 中国电机工程学报, 2005,25,(13): 101-104. doi: 10.3321/j.issn:0258-8013.2005.13.019GUO Xin, ZHENG Chu-guang, LÜ Nai-xia. Density functional theory study of adsorption of mercury and mercury over the CaO(001) Surface of cluster model[J]. Proc CSEE, 2005, 25(13): 101-104. doi: 10.3321/j.issn:0258-8013.2005.13.019
王鹏, 吴江, 任建兴. 飞灰未燃尽碳对吸附烟气汞影响的试验研究[J]. 动力工程学报, 2012,32,(4): 332-337. doi: 10.3969/j.issn.1674-7607.2012.04.012WANG Peng, WU Jiang, REN Jian-xing. Experimental study on the effect of unburned carbon from fly ash on mercury adsorption in flue gas[J]. Chin J Power Eng, 2012, 32(4): 332-337. doi: 10.3969/j.issn.1674-7607.2012.04.012
王军, 蒋建国, 隋继超. 垃圾焚烧飞灰基本性质的研究[J]. 环境科学学报, 2006,27,(11): 141-145. WANG Jun, JIANG Jian-guo, SUI Ji-chao. Study on the basic properties of waste incineration fly ash[J]. J Environ Sci-China, 2006, 27(11): 141-145.
孙俊民, 韩德馨, 姚强. 燃煤飞灰的显微颗粒类型与显微结构特征[J]. 电子显微学报, 2001,20,(2): 140-147. doi: 10.3969/j.issn.1000-6281.2001.02.012SUN Jun-min, HAN De-xin, YAO Qiang. Microscopic particle types and microstructure characteristics of coal-fired fly ash[J]. J Electron Micro, 2001, 20(2): 140-147. doi: 10.3969/j.issn.1000-6281.2001.02.012
WILLIAMS R P, RIESSEN A V. Determination of the reactive component of fly ashes for geopolymer production using XRF and XRD[J]. Fuel, 2010, 89(12): 3683-3692. doi: 10.1016/j.fuel.2010.07.031
NURIA L, FRANCESC I, GIANFRANCO P. Adsorption of Cu, Pd, and Cs Atoms on Regular and Defect Sites of the SiO2 Surface[J]. J Am Chem Soc, 2017, 121(4): 813-821.
FERULLO R M, GARDA G R, BELELLI P G. Deposition of small Cu, Ag and Au particles on reduced SiO2[J]. J Mol Struct (Theochem), 2006, (769): 217-223.
吕鑫.簇-表面类比: 金属氧化物簇模型探讨[D].厦门: 厦门大学, 2004.LÜ Xin. Cluster-Surface Analogy: Discussion on Metal Oxide Cluster Model[D]. Xiamen: Xiamen University, 2004.
MUKHOPADHYAY S, SUSHKO P V. Correlation between the atomic structure, formation energies, and optical absorption of neutral oxygen vacancies in amorphous silica[J]. Phys Rev B, 2005, 71(23): 5204.
KRISHNAN R, BINKLEY J S, SEEGER R. Self-consistent molecular-orbital methods-A basis set for correlated wave functions[J]. Chem Phys, 1980, 72(1): 650-654.
WEIGEND F, AHLRICHS R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn:Design and assessment of accuracy[J]. Phys Chem Chem Phys, 2005, (7): 3297-3305.
GAUSSIAN 09, REVISIOND.01, FRISCH M J, TRUCKS G W, SCHLEGEL H B. Gaussian, Inc., Wallingford CT, 2016.
LU T. Multiwfn:A multifunctional wavefunction analyzer[J]. J Comput Chem, 2012, 33(5): 580-592. doi: 10.1002/jcc.v33.5
TANG T H, CUI Y P. A theoretical study of some X-H-π hydrogen-bonded complexes using the theory of atoms in molecules[J]. Can J Chem, 2011, 74(6): 1162-1170.
BADER R F. A quantum theory of molecular structure and its applications[J]. Chem Rev, 1991, 91(5): 893-928. doi: 10.1021/cr00005a013
CREMER D, KRAKA E. Chemical bonds without bonding electron density-Does the difference electron density analysis suffice for a description of the chemical bond[J]. Angew Chem, 1984, 23(23): 627-628.
ESPINOSA E, ALKORTA I, ELGUERO J. From weak to strong interactions:A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X-H F-Y systems[J]. J Chem Phys, 2002, 117(12): 5529-5542. doi: 10.1063/1.1501133
董兰, 蒋树斌, 郑申声. Ben(n=1~6)团簇吸附H2的密度泛函研究[J]. 原子能科学技术, 2010,44,(12): 1414-1419. DONG Lan, JIANG Shu-bin, ZHENG Shen-sheng. Density functional theory for hydrogen molecule adsorption of Ben(n=1~6) clusters[J]. Atomic Energy Sci Technol, 2010, 44(12): 1414-1419.
叶天旭, 徐永强, 张予辉. Co/MoS2催化剂加氢脱硫活性的量子化学研究[J]. 中国石油大学学报(自然科学版), 2005,29,(4): 119-121. doi: 10.3321/j.issn:1000-5870.2005.04.028YE Tian-xu, XU Yong-qiang, ZHANG Yu-hui. Quantum chemistry study on hydrogenation desulfurization activity of Co/MoS2 catalyst[J]. J Chin Univ Pet(Nat Sci Ed), 2005, 29(4): 119-121. doi: 10.3321/j.issn:1000-5870.2005.04.028
JACOBSEN H J. Localized-orbital locator (LOL) profiles of transition-metal hydride a[J]. Can J Chem, 2009, 87(7): 965-973. doi: 10.1139/V09-060

图 3 SiO2-AsO各吸附构型的拓扑分析图
Figure 3 Topological analysis diagram of SiO2-AsO of various configurations

图 4 SiO2-AsO各吸附构型的LOL填色图
Figure 4 LOL color map of SiO2-AsO of different adsorption configurations

图 5 各吸附构型中相互作用的两原子间对应的BCP处的LOL值
Figure 5 LOL values at corresponding BCPs between two interacting atoms in each adsorbed configuration
表 1 SiO2单体吸附AsO构型的相互作用能
Table 1. Interaction energies between AsO and SiO2 of various adsorption configurations
| Configuration | Eads /(kJ·mol-1) | Configuration | Eads /(kJ·mol-1) | ||
| SiO2-AsO | A1 | -214.14 | SiO2-AsO | C | -386.76 |
| A2 | -248.49 | D | -408.62 | ||
| B1 | -83.04 | E | -427.50 | ||
| B2 | -210.77 | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 SiO2单体吸附AsO构型的吸附热
Table 2. Adsorption heat of AsO on SiO2 with different configurations
| Configuration | Hads /(kJ·mol-1) | Configuration | Hads /(kJ·mol-1) | ||
| SiO2-AsO | A1 | 225.08 | SiO2-AsO | C | 376.52 |
| A2 | 250.65 | D | 404.94 | ||
| B1 | 84.00 | E | 432.04 | ||
| B2 | 217.07 | ||||
下载: 导出CSV
表 3 吸附构型中Si-As键临界点(BCP)的拓扑分析性质
Table 3. Topological parameters of the BCP of Si-As bonds in various adsorption configurations
| Configuration | Bond | ρ | ▽2ρ | G | V | H | |V|/G | Mulliken |
||
| As/O | △ | |||||||||
| SiO2-AsO | A1 | Si1-As8 | 0.242 | -0.013 | 0.085 | -0.154 | -0.204 | 2.12 | 0.150 | -0.322 |
| A2 | Si1-O9 | 0.249 | 0.342 | 0.089 | -0.183 | -0.344 | 2.15 | -0.831 | -0.359 | |
| B1 | Si8-As15 | 0.210 | 0.145 | 0.029 | -0.060 | -0.048 | 2.07 | 0.329 | -0.143 | |
| Si1-As15 | 0.211 | 0.134 | 0.029 | -0.061 | -0.047 | 2.10 | 0.329 | -0.143 | ||
| B2 | Si1-O16 | 0.242 | 0.385 | 0.074 | -0.153 | -0.203 | 2.08 | -0.793 | -0.321 | |
| Si8-O16 | 0.243 | 0.387 | 0.072 | -0.151 | -0.201 | 2.09 | -0.794 | -0.322 | ||
| C | O2-As9 | 0.290 | 0.359 | 0.104 | -0.229 | -0.565 | 2.20 | 0.947 | 0.475 | |
| D | O11-As15 | 0.321 | 0.406 | 0.135 | -0.348 | -0.733 | 2.58 | 0.954 | 0.482 | |
| E | O5-As27 | 0.323 | 0.343 | 0.152 | -0.419 | -0.665 | 2.76 | 1.043 | 0.571 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们