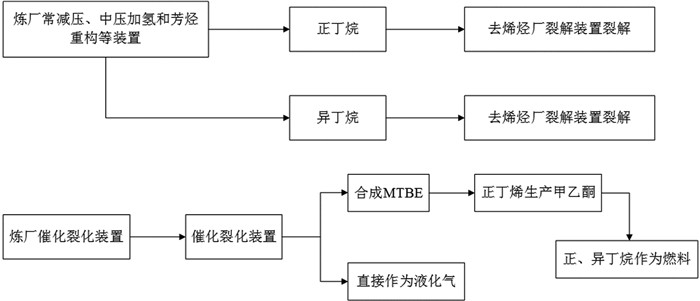
 图1
炼化企业C4烃加工利用现状
Figure1.
Current situation of exploitation of C4 hydrocarbons from refinery enterprises
图1
炼化企业C4烃加工利用现状
Figure1.
Current situation of exploitation of C4 hydrocarbons from refinery enterprises
引用本文:
周庆祥, 刘洋, 柯明. 异丁烷直接脱氢制异丁烯的催化剂的研究进展[J]. 化学通报,
2017, 80(9): 835-844.

Citation: Zhou Qingxiang, Liu Yang, Ke Ming. Research Progress of Catalysts for Isobutane Direct Dehydrogenation to Isobutene[J]. Chemistry, 2017, 80(9): 835-844.
Citation: Zhou Qingxiang, Liu Yang, Ke Ming. Research Progress of Catalysts for Isobutane Direct Dehydrogenation to Isobutene[J]. Chemistry, 2017, 80(9): 835-844.
异丁烷直接脱氢制异丁烯的催化剂的研究进展
English
Research Progress of Catalysts for Isobutane Direct Dehydrogenation to Isobutene
Abstract:
The differences between isobutane direct dehydrogenation and oxydehydrogenation were summarized from the perspective of thermodynamics and reaction mechanism. The catalysts for the direct dehydrogenation of isobutane to isobutene, including PtSn-、CrOx-、GaOx-base and metal sulfide catalyst, are reviewed in terms of the nature of active center, deactivation-regeneration, the promoters and carriers. The advantages and disadvantages of the catalysts and the current research direction were analyzed. Other types of dehydrogenation catalysts were summarized, including Mo/MgAl2O4, carbon-based material, Co-, Ni2P-, VOx-based catalysts, etc. The problems of current dehydrogenation technology and the future research direction were analyzed.
-
Key words:
- Isobutane
- / C4 hydrocarbons
- / Dehydrogenation
- / Isobutene
- / Catalyst
-
C4烃在化工、燃料等方面用途广泛,但是目前国内对C4烃的利用率不足(C4烃加工利用现状如图 1所示[1])。C4烃中的异丁烯作为重要的精细化工原料,能够合成多种高附加值的化工产品,如图 2所示[2]。随着当前全球烯烃市场的迅猛发展,以及异丁烯下游产品的不断开发和利用,使得世界烯烃需求量不断攀升。通常,获得异丁烯的主要技术路线包括蒸汽裂解、催化裂化(FCC)以及催化脱氢。前两种技术获得的产物是混合物,不能有效地获得高纯异丁烯。利用低碳烷烃脱氢工艺生产的目标烯烃选择性高,因此,催化脱氢技术是生产异丁烯的有效替代路线,具有巨大的工业应用价值[3~5]。
图1
炼化企业C4烃加工利用现状
Figure1.
Current situation of exploitation of C4 hydrocarbons from refinery enterprises
 图2
异丁烯衍生物产品链
Figure2.
Product chain of isobutene and its derivative
图2
异丁烯衍生物产品链
Figure2.
Product chain of isobutene and its derivative
1 异丁烷脱氢制异丁烯反应机理
1.1 异丁烷脱氢制异丁烯工艺
异丁烷脱氢制异丁烯工艺分为直接脱氢和氧化脱氢两类,其中,直接脱氢反应是强吸热过程,需要高温下进行,能耗高,但是直接脱氢工艺相对成熟,已商业化的脱氢催化剂性能稳定;而氧化脱氢与直接脱氢不同,不受热力学限制,能够在400℃下进行,这是因为氧化脱氢反应过程中生成的氢气经氧化剂(如O2、CO2等)燃烧掉,使得烷烃完全转化。但是,氧化脱氢存在一大缺陷,即原料中烷烃的过度氧化,难以满足高的烯烃选择性。目前,氧化脱氢反应中常用的氧化剂为CO2,较为温和,能够在脱氢条件下经逆向水煤气变换从而有效地消耗掉H2,而不足以氧化烃类物质,但是CO2在催化剂上的竞争吸附会影响催化剂的脱氢性能[6, 7]。因此,需要寻找一种更为稳定的氧化剂和良好的催化剂是推进氧化脱氢实现工业化的重中之重。
1.2 异丁烷脱氢制异丁烯反应机理
脱氢反应已被研究了近一个世纪,其准确的反应机理仍富有争议,催化剂的类型不同,其对应的催化脱氢机理也有所差异。1934年提出的Horiuti-Polanyi机理被普遍用于解释脱氢反应过程,该机理符合Langmuir-Hinshelwood动力学,认为催化剂表面的活性位是一致的,具体包含四个步骤:烷烃的解离吸附;C-H键断裂产生第二个氢原子;氢分子的形成;氢分子和烯烃分子的脱附。普遍认为,C-H键断裂和烷烃分子的解离吸附是脱氢反应的速控步骤[8, 9]。对于金属氧化物催化剂,如Cr基催化剂,其脱氢反应机理有所不同,除了金属粒子外,氧化物中的氧原子也参与催化反应。此机理认为,速控步骤是烷烃分子的解离吸附,或者β-H消去[10, 11]。
总之,大部分实验结论已证实,轻质烷烃脱氢机理符合Horiuti-Polanyi机理,其速控步骤为烷烃分子的解离吸附或第二个C-H键解离。
2 异丁烷直接脱氢制异丁烯的催化剂
异丁烷直接脱氢反应是一个受平衡限制且高度吸热的过程,反应条件相对苛刻,因此要求催化剂能够限制因烧结、挥发、水热再生等因素所造成的不可逆失活以及形貌或相态的改变。图式1为异丁烷脱氢反应网络图[12]。
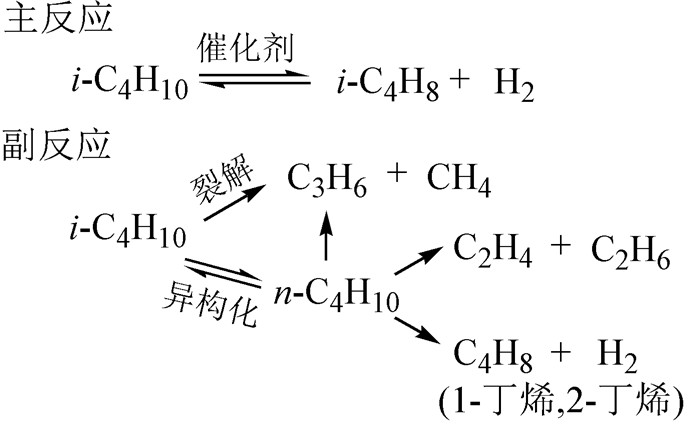 图式1
异丁烷脱氢反应网络图
Scheme1.
Reaction network of isobutane dehydrogenation
图式1
异丁烷脱氢反应网络图
Scheme1.
Reaction network of isobutane dehydrogenation
烯烃分子与其对应的烷烃分子相比具有更高的反应活性,容易发生副反应或二级反应(如氢解反应、裂化反应、异构化反应),这些副反应的发生需要在特定的催化活性中心(如团聚的、缺陷多的金属活性中心)上进行。此外,由于烷烃和烯烃中的C-H键比C-C键更活泼,因此,适合的催化剂更青睐于断裂C-H键以抑制副反应的发生。异丁烯因其π键上的高电子密度而被认为是Lewis碱且具有亲核性,因此,弱酸中心有利于异丁烷脱氢反应的进行[13]。总之,选择酸性适中的载体和活性良好的金属组分是设计一种合理的脱氢催化剂的核心,而脱氢催化剂的合理设计是获得高烯烃收率的关键。
用于异丁烷直接脱氢的催化剂主要分为两类:贵金属催化剂和金属氧化物催化剂。Pt基催化剂是唯一一种已工业化的贵金属脱氢催化剂,CrOx是金属氧化物脱氢催化剂的典型代表。目前,已工业化的和文献中广泛报道的直接脱氢催化剂见表 1。
 表 1
异丁烷直接脱氢制异丁烯催化剂汇总
Table 1.
Summary of catalysts for isobutane direct dehydrogenation to isobutene
表 1
异丁烷直接脱氢制异丁烯催化剂汇总
Table 1.
Summary of catalysts for isobutane direct dehydrogenation to isobutene
类别 特点 举例 目前研究方向 PtSn基负载型催化剂 易活化C-H键,脱氢活性高,可以通过控制活性Pt物种尺寸以抑制副反应进行,但是易结焦且成本高 Pt-Sn-K/γ-Al2O3[14] 助催化剂、载体的选择以及制备,力求降低成本的同时提高催化剂的活性、稳定性和寿命 CrOx基催化剂 适用于直接脱氢和氧化脱氢,成本低,但是失活迅速,在高温下不稳定,且高价Cr对人体健康和环境保护不利 Cr2O3-K2O/Al2O3[15] 载体、助催化剂的选择以及催化脱氢机理的研究 GaOx基催化剂 适用于直接脱氢和氧化脱氢,含有大量的弱Lewis酸中心,催化剂上稳定存在的β-Ga2O3是脱氢活性中心 Ga2O3/MTS,Ga/Si=0.05[16] 包括载体和助催化剂的研究 金属硫化物催化剂 活性和选择性高,初始收率高于商业化的CrOx和Pt-Sn催化剂,但是几小时后催化剂因硫以H2S形式流失而迅速失活,对装置材料的抗腐蚀性要求较高 CoS/SiO2、FeS/SiO2[68] 提高催化剂的寿命和稳定性的研究 其他 — Mo/MgAl2O4、碳基材料、Co-、Ni2P-、VOx基催化剂等 研究具有高脱氢活性、选择性以及稳定性的新型脱氢催化剂 2.1 Pt-Sn基催化剂
源于催化重整工艺的双功能负载型金属催化剂是由酸性载体负载贵金属组合而成,其中,酸性载体为异构化、加氢裂化和环化反应的进行提供了活性位,而贵金属是催化加氢和脱氢的活性位。为了提高该催化剂的脱氢活性、抑制副反应的发生,必须消除其酸催化功能。
Pt活化C-H键的活性较活化C-C键的高,这是因为在Pt原子表面,除了低配位数的Pt活性位点(如台阶、扭结)能够催化C-C键断裂外,其余Pt活性位点几乎全部能够活化C-H键。由此可知,结焦之类的副反应属于结构敏感型反应,通常发生在团聚的Pt晶簇上[11, 17],而烷烃脱氢反应主要发生在分散的小Pt晶粒上,因此要求Pt基脱氢催化剂上的Pt尽可能地分散,且晶粒度越小越好。图 3为Pt基催化剂脱氢反应网络图。
2.1.1 Pt-Sn基催化剂的失活与再生
造成Pt基脱氢催化剂失活的主要原因有两个:一是催化脱氢的同时发生了副反应,在催化剂表面生成焦炭,覆盖了活性位,造成活性中心数目减少,催化活性降低;二是脱氢和再生均需要在高温下进行,而高温易引起Pt粒子的迁移和团聚[19]。由此可知,可以从引入助剂和改进载体两个方面出发,通过提高Pt粒子的分散度、减小Pt晶粒大小、调节Pt与载体间相互作用从而延缓催化剂的失活。
由于烷烃脱氢过程中不断有焦炭生成,因此Pt基催化剂需要频繁再生。常用的再生方法是将失活的催化剂在O2气氛下燃烧积炭,以及在氯气、氧气气氛下处理(即氧氯化)。其中,氧氯化除了能够烧掉焦炭,还可对团聚的Pt物种进行再分散,因此此法是Pt基催化剂最有效的再生手段。积炭的位置决定了积炭燃烧的温度,如果是Pt粒子上的积炭则需要在相对低温下氧化(~450℃),如果是载体上的积炭则需要高温氧化(>500℃),这是由于载体上积炭的氧化需要消耗从金属转移到载体上的溢流氧。此结论能够通过程序升温氧化(TPO)和透射电子显微镜(TEM)手段得以证实[20]。
2.1.2 助催化剂
Pt基催化剂中引入合适的助催化剂(下文简称“助剂”),能够有效抑制脱氢过程中的副反应,从而提高产物烯烃选择性和催化剂稳定性。工业上使用最广泛的助剂是Sn,它既是几何型助剂,也是电子型助剂。一方面,Sn能够减小Pt晶粒尺寸,增大Pt粒子分散度,减少氢解和积炭反应所需的较大的Pt晶粒;另一方面,Sn提供电子给Pt,占据Pt原子的5d轨道,削弱了Pt与烯烃分子间的相互作用,促使产物烯烃脱附,从而抑制氢解和积炭,此外,Snn+物种所带的正电荷能够改变Pt的电子云密度。Sn的存在还能够降低载体酸性,促使焦炭从金属表面迁移到载体上,延缓金属烧结[20~26]。
除了Sn外,Pt基催化剂常用的助剂还有碱金属和碱土金属。其中,碱金属(如Li、Na、K)能够中和载体上的酸性位,抑制异构化等副反应发生,提高催化剂的脱氢活性和稳定性[27, 28];而碱土金属能够改善金属功能,增强金属-载体间相互作用[14]。此外,文献报道中还研究了其他用作Pt基脱氢催化剂助剂的金属,如铜[29]、锌[30]、镓[31, 32]、锗[33]、铟[34]等。其中,Zn与Sn的作用类似,能够改善Pt基催化剂的几何和电子特性,抑制积炭、异构化等副反应进行[23, 30, 35]。
2.1.3 载体
异丁烷脱氢反应是吸热过程,需要较高的反应温度,因此烷烃脱氢催化剂载体必须具有足够的耐热稳定性。此外,载体的酸碱性以及其比表面积和孔径分布等结构性质也是决定催化性能的重要因素。载体的酸碱性不仅影响活性物种的结构及其分散度,还影响反应物与反应中间体在催化剂表面上的停留时间[36]。负载型PtSn双金属催化剂中Sn的还原程度与催化剂制备方法、锡前驱体种类以及载体类型紧密相关,载体的组成又会影响金属粒子的稳定性,该稳定性表现在金属粒子的烧结以及载体上的结焦方面。早前的研究已经证实,使用如K-L分子筛[37, 38]、碱掺杂的氧化铝[39]、尖晶石[40]、焙烧后的水滑石[41]等无酸性的载体能够减缓催化剂结焦;此外,一些沸石分子筛[42~44]、氮化处理的镓铝磷酸盐[45]等材料均可用作Pt基催化剂的载体。工业使用的Pt基脱氢催化剂的载体是高表面积的氧化铝,Pt-Sn/Al2O3催化剂表面主要为Lewis酸中心,几乎不存在Brønsted酸中心,而且Al2O3载体热稳定性好,机械强度高,能够很好地分散Pt纳米粒子。但是,大部分的氧化铝载体呈酸性,容易发生结焦等副反应,因此,需要加入助剂来调控其酸性。
尖晶石型结构具有良好的热稳定性、弱酸性以及疏水性,能够很好地适用于脱氢反应。如ZnAl2O4或MgAl2O4尖晶石能够增强金属-载体间相互作用,抑制Pt晶粒烧结[39]。Miguel等[46]对比研究了不同载体对负载型PtSn双金属脱氢催化剂稳定性及选择性的影响。分析发现,MgAl2O4和涂层法制备的复合氧化铝载体负载能够增强金属物种间的相互作用,阻碍金属物种团聚,因此表现出较高的脱氢活性、选择性和稳定性。
水滑石呈碱性,可用作Pt基脱氢催化剂的载体,能够很好地抑制结焦,但是烯烃选择性较差,这是因为烷烃反应物在Pt粒子上容易二次吸附,产生焦炭,导致催化剂失活,因此需要在载体中引入Ga、Ge、In等氧化物助剂以改善其催化性能。Bell等[31, 32]将GaOx引入水滑石载体中,发现Ga改性的Pt基催化剂中,Ga破坏了载体表面的Brønsted酸中心,抑制了副反应发生。此外,在Pt/Mg(Ga)(Al)O催化剂高温还原期间,Pt粒子表面生成的H原子溢流到载体上,促使载体上的Ga3+还原为Ga0,继而与Pt纳米粒子相互作用形成GaPt合金。此类双金属合金具有几何和电子效应,促使烷烃反应物解离吸附,同时又促进产物烯烃脱附。
沸石分子筛兼有Brønsted酸中心和Lewis酸中心,具有分布良好的孔道结构,利于控制反应过程中的质量传递和热量扩散。ZSM-5分子筛常用于负载PtSn双金属并用于催化丙烷脱氢反应,但是,纯ZSM-5的酸度、抗积炭能力和稳定性欠佳。Nawaz等[47]对比了不同分子筛负载的PtSn催化剂(Pt-Sn/slit-SAPO-34、Pt-Sn/ZSM-5) 对轻质烷烃脱氢反应的催化性能。分析表明,由于Pt-Sn/ZSM-5孔道大小适合原料分子进入,故脱氢活性较高,但由于表面酸性较强,容易裂化;而slit-SAPO-34具有良好的择形选择性和较高的表面扩散率,因此Pt-Sn/slit-SAPO-34催化剂表现出高烯烃选择性和热稳定性。
Escribano等[48]认为惰性载体(如SiO2、C)能够削弱Sn与载体间相互作用,从而增强Pt-Sn间相互作用,促使PtSn合金生成。但是,过量的PtSn合金会降低Pt基催化剂的脱氢活性,因此他们通过引入CeOx限制Sn物种的还原,保证少量PtSn合金的存在,实现在不影响脱氢活性的同时有效地抑制氢解和结焦。Ohta等[49]以ZnO-Cr2O3为载体制备了PtSn/ZnO-Cr2O3催化剂用于异丁烷低温脱氢,研究了Sn含量对该催化剂脱氢性能以及抗积炭能力的影响。
Pt-Sn基催化剂在工业应用方面仍存在高成本和积炭失活两方面的问题,因此,目前对Pt-Sn基催化剂重点研究包括助剂、载体的选择以及制备方法,力求降低成本的同时能够提高催化剂的活性、稳定性和使用寿命。本节介绍的部分Pt基催化剂性能见表 2。
表 2
Pt基催化剂催化性能参数汇总
Table 2.
Summary of the catalytic data of the Pt-based catalysts
催化剂 制备方法 反应温度/℃ 空速 进料组成 转化率/% 选择性/% 文献 2(wt)% Pt-1(wt)%
Sn/CeO2/C共浸渍 500 WHSV=6h-1 H2/i-C4H10=6 37~20 90~100 [48] Pt-Sn/MA-P1 真空等体积浸渍 580 1200mL i-C4H10/gCat. H2/i-C4H10=1:1 55.8~53.7b 65b [50] PtSn-La/Al2O3 分步浸渍 590 10mL i-C4H10/gCat. H2=10mL/min
i-C4H10=5mL/min49.4c 97c [51] PtSnK/γ-Al2O3d 分步浸渍 590 GHSV=3000h-1 H2/i-C4H10=2 29.0 94 [14] Pt-Sn/slit-SAPO-34e 共浸渍 585 WHSV=5h-1 H2/i-C4H10=0.2 40.1~16.0 84 [47] a0.2(wt)% Pt,且Sn/Pt=0.5,并标明了初始化转化率及选择性;b分别为始末转化率和初始选择性;c分别为反应220min时刻的转化率和选择性;d催化剂配比为0.5(wt)% Pt,0.5(wt)% Sn,0.3(wt)%K,且标明了反应6h后的转化率和选择性;e催化剂配比为0.5(wt)% Pt,1(wt)% Sn,并标明了初始转化率及反应8h后的选择性。 2.2 CrOx基催化剂
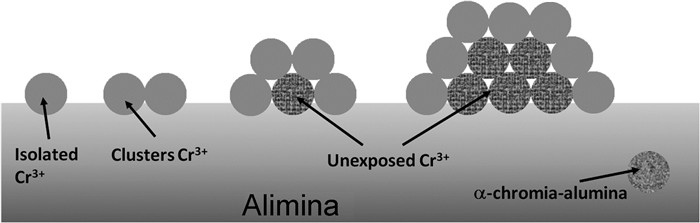
1933年,Frey和Huppke报道了氧化铬具有脱氢活性,不久,CrOx基催化剂首次用于生产烯烃,CrOx/Al2O3脱氢催化剂于1943年实现商业化。CrOx基催化剂既适用于异丁烷直接脱氢,又适用于氧化脱氢,其中,氧化脱氢反应温度为200~400 ℃,直接脱氢反应温度在500℃以上[52, 53]。新鲜的CrOx基催化剂表面已确定的物种包括Cr2+、Cr3+、Cr5+、Cr6+以及α-Cr2O3晶体,具体取决于预处理方式、Cr的负载量和前驱体种类、焙烧温度以及载体性质[54]。如图 4所示,以Al2O3为载体的催化剂表面铬物种分为[55, 56]:(1) 高度分散的Cr6+物种,主要存在于单层金属负载量的催化剂中,以高氧化态形式与载体相互作用;(2) 孤立的Cr3+物种,部分扩散到Al2O3骨架中;(3) 构成小晶簇的Cr3+物种,存在于中等或高金属负载量的催化剂中;(4)α-Cr2O3,存在于高金属负载量的催化剂中。
烷烃分子的吸附位点在Cr-O上。研究发现,高温下,在异丁烷或正丁烷存在的条件下,催化剂表面的Cr6+在脱氢反应初期转变为拟八面体的Cr3+,Cr5+被烷烃分子还原为Cr3+。因此多铬酸盐的催化位点比铬酸盐更容易减少。在烷烃脱氢过程中,CrOx/Al2O3催化剂上配位不饱和的Cr3+通过吸附气相中的烷烃分子从而转化为大体积的稳定的Cr3+。利用波谱表征技术推断出Cr3+分3类:由表面Cr5+或Cr6+还原得到的Cr3+、固定在Al2O3表面上的孤立的Cr3+、无定型Cr3+晶簇[57]。
Fang等[59]利用XRD、SEM、XPS、H2-TPR、TPO等多种表征手段研究了Cr-Al2O3催化剂对异丁烷脱氢的催化特性以及失活机理。分析结果显示,在新鲜的和再生后的催化剂中包含Cr3+和Cr6+两种铬物种,而使用中的催化剂则包含Cr3+和Cr2+两种铬物种。Cr3+无法被H2还原,却能够被烷烃或CO还原为Cr2+,说明Cr3+和Cr2+为异丁烷脱氢反应的活性中心。
综上所述,孤立的以及配位不饱和的Cr3+物种决定了催化剂的脱氢活性,而其他铬物种则用于补充反应进行中耗尽的Cr3+。
2.2.1 CrOx基催化剂的失活与再生
脱氢循环过程中引起催化剂活性改变的原因包括Cr6+还原为Cr3+和催化剂结焦。CrOx基催化剂的脱氢活性与还原得到的拟八面体的Cr3+数目有关。反应过程中表面Cr6+物种易被还原为Cr3+物种,即表面Cr6+物种是Cr3+物种的前驱体,因此表面Cr6+的数目随着反应-再生循环次数的增加而减小[60]。Cr3+与Al3+具有相近的离子半径和电荷,因此经高温灼烧再生后的CrOx基催化剂因焦炭燃烧释放的热量促使活性铬物种进入Al2O3载体的八面体结构空隙中,形成一种Cr2O3-Al2O3尖晶石,稳定地存在于氧化铝载体中。但是,Cr2O3-Al2O3尖晶石无脱氢活性,大量Cr2O3-Al2O3尖晶石会造成催化剂失活。同时,催化剂表面可接触的活性中心随着反应的进行可能发生团聚、被焦炭覆盖,造成催化剂表面积不断降低。因此与Pt基催化剂类似,CrOx基催化剂容易因积炭而失活,需要定期高温烧炭再生[61]。
2.2.2 助催化剂
多数碱金属(如K、Na、Cs、Rb)常用作铬基催化剂的助剂,不仅能够稳定载体结构、降低氧化铝载体表面酸性以抑制裂化等副反应发生,还能够增加活性Cr物种,并提高其分散度。Rombi等[55]研究发现,K能够改变表面化学性质和催化行为,随着K含量的增加(0~1.8(wt)%),催化剂表面酸性明显降低;以α-Cr2O3形式存在的Cr3+物种脱氢活性低于不饱和Cr3+物种,而α-Cr2O3则随着K含量的增大而减少。此外,Cr6+物种浓度随着K含量的增加而增加,说明K的引入能够改善催化剂的酸度和Cr6+/Cr3+比值。
Lang等[62]利用UV-Vis、XPS、H2-TPR、NH3-TPD等表征手段研究了Ni对Cr/Al脱氢催化剂性能的影响。结果表明,Ni助剂的作用主要有:(1) 促进低聚态铬物种的形成,影响载体表面Cr物种的价态,促使Cr6+还原为Cr3+;(2) 减少催化剂表面弱酸中心数目,降低总酸量;(3) 削弱Cr与载体间的相互作用,同时,Ni-Cr之间协同作用促进了丙烷脱氢反应的进行。
2.2.3 载体
载体能够影响CrOx基催化剂脱氢活性和表面Cr物种分散度;而且,载体与活性组分之间的相互作用对催化剂活性和选择性也有显著影响。商业使用的CrOx基催化剂载体为氧化铝,但是,氧化铝最大的缺陷是其表面酸性有利于催化裂化和结焦反应的发生,造成活性和选择性均降低,因此,通常要求掺杂碱金属来调变其表面酸性。
ZrO2的酸度低、热稳定性高,是一种潜在的良好的CrOx基催化剂载体。Rossi等[63]将CrOx/ZrO2催化剂与CrOx/γ-Al2O3和CrOx/SiO2作对比,研究各自的异丁烷脱氢活性。结果发现,裂化的活性位是Brønsted酸中心,而ZrO2载体具有极少的Brønsted酸中心,因此在相同反应条件下CrOx/ZrO2的平均转换频率高于CrOx/γ-Al2O3和CrOx/SiO2。Korhonen等[64]对比了ZrO2、Al2O3、ZrO2-Al2O3三种载体对CrOx基催化剂脱氢性能的影响,利用原位DRIFTS、Raman等手段研究发现,ZrO2沉积在Al2O3中能够降低载体的Lewis酸度,抑制催化剂结焦,但脱氢活性较低,而CrOx/ZrO2催化剂脱氢活性最高。
Udomsak等[65]分别采用改性的溶胶-凝胶法、机械混合法、正硅酸乙酯嵌入晶体钛酸盐焙烧法制备了三种SiO2-TiO2混合氧化物载体(Si/Ti=0.36),结果表明,利用焙烧法制备的CrOx/TiO2-SiO2催化剂活性与工业化CrOx基催化剂最接近,异丁烯初始收率可达60%。
介孔材料具有比表面积高、孔径分布均一以及孔道尺寸大等特点,常常用作催化剂载体。Dogan等[66]利用水热合成法和浸渍法制备了Cr/MCM-41催化剂,并研究了其失活原因,表征结果发现,催化剂表面具有高度分散的Cr物种,而且含有大量的Cr6+,其中四配位的Cr(Ⅳ)O4物种形成的单铬酸盐最具异丁烷脱氢活性。但是,四配位的Cr(Ⅳ)O4物种在反应过程中易被还原为无催化活性的八面体配位的Cr(Ⅲ)O6物种和Cr2O3晶相,造成催化剂失活。此外,MCM-41载体的水热稳定性仍待提高。
Cr2O3-K2O/Al2O3催化剂是低碳烷烃脱氢反应中使用较多的CrOx基催化剂,具有高活性和选择性。但是,该催化剂在高温下不稳定,低温下活性较差,需频繁再生;而且高价态的Cr物种对人体健康和环保不利,所以此类催化剂的使用受到了限制。部分CrOx基催化剂性能见表 3。
表 3
CrOx基催化剂催化性能参数汇总
Table 3.
Summary of the catalytic data of the CrOx-based catalysts
催化剂 制备方法 反应温度/℃ 空速 原料组成 转化率/% 选择性/% 文献 3(wt)% Cr-MCM-41 原位水热合成 600 WHSV=26 h-1 i-C4H10=20 mL/min 27a - [66] 15(wt)% Cr-Al2O3b 浸渍 600±5 - 纯i-C4H10 63~30 90 [59] CrOx/nZrO2-SiO2c 等体积浸渍 540 GHSV=540 h-1 i-C4H10=270 mL/min
N2=1530 mL/min- >96 [67] Cr10Zr90Od 共浸渍 550 0.24 g·s·mL-1 i-C4H10/H2=40:60 36 84 [68] K2O-Cr2O3-B2O3/γ-Al2O3e 等体积浸渍 560 400 h-1 H2/i-C4H10=6 57.1~36.4 92.6~93.2 [69] a反应15min时达到最大值;b标明了其始末转化率及反应750min时的选择性;c n=1.0~2.0 (wt)%,催化活性为反应15min时的选择性;d标明了反应9min时的初始转化率及选择性;e2 (wt)% K2O,12 (wt)% Cr2O3,1 (wt)% B2O3,并分别标明了反应0.5h和8h时的转化率和选择性。 2.3 GaOx基催化剂
近几年,氧化镓基脱氢催化剂得到广泛关注,研究发现,此类催化剂存在稳定的β-Ga2O3物种,被认为是烷烃脱氢的活性中心,该物种具有单斜晶体结构,含有大量的弱Lewis酸中心[70]。异丁烷在GaOx基催化剂表面的双吸附位上发生异裂解离反应,H+吸附在Ga+(Lewis酸中心)上,而C4H9+吸附在相邻的氧活性位上。
Nesterenko等[71]以MCM-41型介孔SiO2(记作MTS)为载体,制备了Ga-MTS、Fe-MTS催化剂,并与Si-MTS、Al-MTS作对比,比较脱氢活性及稳定性。结果表明,四种催化剂中,Ga-MTS催化剂表现出最高的异丁烷脱氢活性,分析其原因为,Ga-MTS催化剂具有高度分散的氧化镓物种,450℃、H2气氛下,Ga3+易被还原为Gaδ+(δ < 2),还原性Gaδ+能够断裂C-H键,促使H2和烯烃快速重组脱附。Li等[72]考虑到金属镓资源稀少且价格昂贵,将ZnO作为载体通过机械混合制备了一系列ZnO-Ga2O3复合金属氧化物催化剂,在降低了Ga2O3用量的同时,ZnO与Ga2O3之间的协同作用增强了该催化剂对异丁烷的吸附力。此外,ZnO上负载的Ga2O3为催化剂提供的Lewis酸中心促进了C-H键异裂,提高了异丁烷转化率。异丁烷脱氢活性由高到低为:ZnO-Ga2O3>ZnO>Ga2O3。当nZnO/nGa2O3=5时,异丁烯收率高达55.1%。Han等[73]合成了一种具有独特的、集大/中/微孔于一身的ZSM-5中空纤维载体,负载了Ga2O3脱氢活性组分,与常规ZSM-5载体相比,ZSM-5分子筛能够有效地增强催化剂的双功能性,提高活性位的可接触性和分散度。
2.4 金属硫化物催化剂
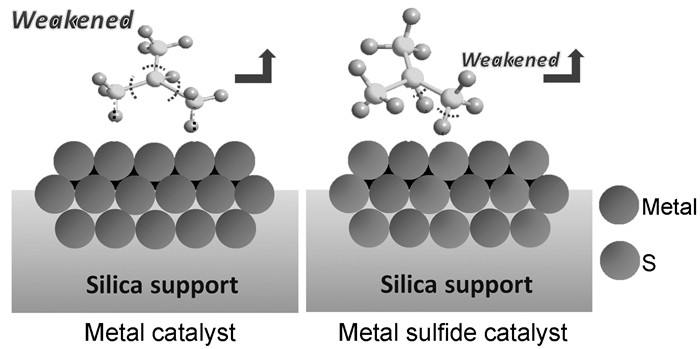
Wang等[74~78]较为系统而全面地研究了金属硫化物催化剂对低碳烷烃脱氢反应的催化性能,以SiO2为载体负载金属硫化物(如MnS、FeS、CoS、NiS、CuS、ZnS和MoS),用于异丁烷脱氢制异丁烯。结果发现,经硫化处理的催化剂其脱氢活性明显比相应的氧化物活性高,这是由于硫的引入增大了M-S键的空间位阻,削弱了异丁烷分子上C原子与金属活性位的相互作用,从而抑制了因C-C键断裂而引起的裂化反应,提高了脱氢性能,如图 5所示。异丁烯收率从大到小顺序如下:CoS/SiO2>FeS/SiO2> NiS/SiO2>Cu2S/SiO2>MnS/ SiO2>MoS2/SiO2>Cr2O3/SiO2>Pt-Sn/SiO2>ZnS/SiO2[76]。他们还以Fe、Co、Ni金属硫化物催化剂为例,分析了S的作用。一方面,S能够稀释团聚的金属物种,降低催化剂氢解活性,抑制C-C键断裂(几何效应);另一方面,S能够促使烯烃产物脱附,提高烯烃选择性(电子效应)。虽然此类催化剂初始收率高于商业使用的CrOx和Pt-Sn基催化剂,但是几小时后由于硫以H2S形式流失而造成催化剂快速失活[77]。
2.5 其他
众所周知,钼基催化剂安全且价格相对低廉,广泛用作烷烃氧化脱氢催化剂。最近,Wang等[79]研制出能够用于异丁烷直接脱氢的Mo/MgAl2O4催化剂,并研究了Mo负载量的最佳值和异丁烷脱氢活性位,分析推测Mo4+为催化脱氢活性中心,当Mo含量为5(wt)%时,该催化剂表现出高脱氢活性和稳定性,异丁烯收率达到35%。
Hao等[80]研究发现,椰壳型活性炭具有良好的异丁烷脱氢活性,活性中心包括表面含氧官能团以及碳本身。但是,碳材料的耐热稳定性有限,因此其再生技术仍待研究。
Wang等[81]还考察了载体(Al2O3、SiO2、MgAl2O4)、焙烧温度(560℃和800℃)以及助剂(S、Sn、Cu、Cl)对Co基催化剂脱氢性能的影响,发现载体和焙烧温度能够调变催化剂的酸性质、Co的分散度以及载体-Co物种间相互作用,从而影响对异丁烷和异丁烯的吸附-脱附性质。对于Al2O3和MgAl2O4负载的Co基催化剂,高焙烧温度有利于提高异丁烯选择性;但是对于Co/SiO2催化剂,高温焙烧后易生成CoSiO4,导致活性位减少、脱氢活性下降。助剂的作用主要是抑制活性金属团聚、增强对H原子的结合力以促使平衡向正向移动。研究发现,助剂Sn和Cu能够抑制氢解反应,但是会降低异丁烷转化率;此外,经Cl改性的Co基催化剂脱氢活性有所降低。
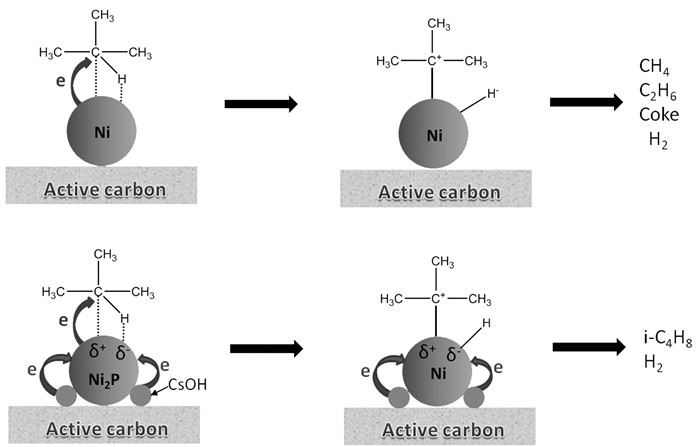
许燕丽等[82]发现磷化镍对异丁烷脱氢制异丁烯具有催化活性,当nNi/nP=1时,异丁烯选择性可达80%。Xu等[83, 84]还发现,Cs改性的Ni2P/AC催化剂具有较高的脱氢活性,这是因为Cs能够提高Ni2P的分散度,促进电子从Cs传递给Ni,降低酸中心数目和酸强度,如图 6所示。Ni2P/AC催化剂反应4 h后,异丁烷转化率达24%,异丁烯选择性>93%。
早在1980年,钒系催化剂已用于烷烃氧化脱氢反应[85]。对VOx基催化剂,研究者认为还原态的VOx能够催化丁烷脱氢,而位于四面体配位中的孤立的V5+物种能够催化丙烷脱氢,但是仍缺乏对异丁烷非氧化脱氢活性位的证实以及不同VOx物种对该反应影响的研究。近来,Tian等[86]制备了10(wt)% V-K/γ-Al2O3,分析发现,还原态的VOx物种,即处在拟四面体配位的低聚态VOx物种(V3+和V4+)是异丁烷脱氢的活性中心。由于Brønsted酸中心容易造成异丁烷裂化,而且烯烃产物易与Brønsted酸中心作用发生二次反应,因此,催化剂的Lewis酸中心与Brønsted酸中心数目的比值直接影响到异丁烯选择性。Rodemerck等[87]认为,VOx物种的聚合程度以及表面酸性对催化剂性能至关重要,二者造成异丁烯骨架异构,从而影响了异丁烯选择性。
上述脱氢催化剂异丁烷脱氢活性参数详见表 4。
表 4
讨论的其他类型脱氢催化剂催化性能参数汇总
Table 4.
Summary of the catalystic data of other types dehydrogenation catalysts discussed
催化剂 制备方法 反应温度/℃ 空速 原料 转化率/% 选择性/% 收率/% 反应时间/h 文献 Mo/MgAl2O3 等体积浸渍 560 GHSV=54.5 h-1 i-C4H10=2mL/min
N2=12mL/min78a 45a 35a 200 [79] CSACb - 625 WHSV=6000mL·g-1·h-1 i-C4H10=1mL/min
N2=19mL/min70 76 53.2 1 [80] OMC8c 浸渍 620 6000mL·g-1·h-1 i-C4H10=1mL/min
N2=19mL/min38 81 30.8 66.7 [88] 13(wt)% Co/SiO2d 等体积浸渍 560 - i-C4H10=2mL/min
N2=12mL/min71e 97e 61.8e 2 [81] Ni2P/SiO2 浸渍 460 800h-1 i-C4H10=2mL/min
N2=12mL/min~4.6e ~72e 80e 2 [82] 6.5(wt)% Cs-Ni2P/SiO2 等体积浸渍 460 1600h-1 i-C4H10/N2=10/10 6 96 5.8 4 [84] Ni2P/AC 等体积浸渍 560 1200h-1 i-C4H10/N2=1/1 15 89 13.4 4.5 [83] 10(wt)%VOx-K2O/γ-Al2O3 等体积浸渍 590 2250mL·g-1·h-1 i-C4H10/N2=1/1 ~61 ~70 42.5 1.75 [86] 注:a催化剂装填量为4g(80~180 μm),数值为平均值;bCSAC为椰壳形活性炭;COMC8(有序介孔碳)经800℃焙烧2h;d经硫化处理2h;e为初始活性。 3 结语
对于异丁烷脱氢制异丁烯的反应,已商业化的Pt基催化剂成本高,CrOx基催化剂高温下不稳定、易迅速失活且存在环境污染的弊端;此外,VOx、GaOx和MoOx基脱氢催化剂失活太快,金属硫化物催化剂具有较高的活性和选择性,但是对装置材质的抗腐蚀性要求较高。现有的脱氢催化剂随着反应的进行均面临失活的问题,而不同类型的催化剂,其失活机理各有差异,但是,结焦是普遍认为的失活的重要因素。究其原因为,催化剂表面形成的焦炭覆盖了活性位和/或孔道,阻碍反应物分子扩散。减少结焦的措施包括使用几乎没有/不含Brønsted酸中心的载体,引入助剂以中和催化剂上的Brønsted酸中心,加入水蒸气、H2稀释剂或CO2等氧化剂共进料。此外,载体-金属间相互作用也是影响催化剂活性的重要因素。失活后的脱氢催化剂需要循环再生,而再生条件需要严格控制,否则可能导致催化剂的不可逆失活。因此,探寻一种适宜的脱氢催化剂,不仅要考虑其载体的酸性,以调节金属-载体间相互作用,还要考虑活性组分的颗粒大小和分散度。
目前页岩气产品的热潮已经证实天然气成本较原油更具有经济性,因此,发展脱氢等生产烯烃的替代方法对传统的催化裂化等方法极具竞争力。但是,烷烃脱氢催化剂仍存在很大的提升空间,除了进一步降低贵金属催化剂的成本、提高催化剂的热稳定性以及脱氢过程中的能源利用率外,还要努力克服受限的平衡转化率以及高温条件下容易发生的副反应,这些是未来低碳烷烃脱氢制烯烃技术亟待解决的关键问题。
-
-
[1]
李涛. 石油化工技术与经济, 2015, 31(2):1~6. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=syha201502001&dbname=CJFD&dbcode=CJFQ
-
[2]
杨为民. 化工进展, 2015, 34(1):1~9. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hgjz201501001&dbname=CJFD&dbcode=CJFQ
-
[3]
刘莹. 石油化工, 2016, 45(5):630~635. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=syhg201605023&dbname=CJFD&dbcode=CJFQ
-
[4]
王堂博, 王广建, 孙万等. 工业催化, 2016, 24(4):1~6.
-
[5]
栗士瑞. 化工设计通讯, 2016, 42(4):177. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=wgtx201604149&dbname=CJFD&dbcode=CJFQ
-
[6]
Q J Zhu, M Takiguchi, T Setoyama et al. Catal. Lett., 2011, 141(5):670~677. doi: 10.1007/s10562-011-0566-6
-
[7]
J F Ding, Z F Qin, S W Chen et al. J. Fuel Chem. Technol., 2010, 38(4):458~461. doi: 10.1016/S1872-5813(10)60040-0
-
[8]
L Horiuti, M Polanyi. Transac. Faraday Soc., 1934, 30:1164~1172. doi: 10.1039/tf9343001164
-
[9]
R D Cortright, P E Levin, J A Dumesic. Ind. Eng. Chem. Res., 1998, 37(5):1717~1723. doi: 10.1021/ie970917f
-
[10]
S M K Airaksinen, M E Harlin, A O I Krause. Ind. Eng. Chem. Res., 2002, 41(23):5619~5626. doi: 10.1021/ie020371j
-
[11]
J J Sattler, J Ruiz-Martinez, E Santillan-Jimenez et al. Chem. Rev., 2014, 114(20):10613~10653. doi: 10.1021/cr5002436
-
[12]
时海涛. 现代化工, 2016, 36(8):142~145.
-
[13]
阎子峰. 化学进展, 2005, 17(4):651~659. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxjz200504009&dbname=CJFD&dbcode=CJFQ
-
[14]
Y Zhang, Y Zhou, L Wan et al. Fuel Proc. Technol., 2011, 92(8):1632~1638. doi: 10.1016/j.fuproc.2011.04.011
-
[15]
肖锦堂. 天然气工业, 1994, 14(6):64~67. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=trqg404.019&dbname=CJFD&dbcode=CJFQ
-
[16]
N S Nesterenko, O A Ponomoreva, V V Yuschenko et al. Appl. Catal. A, 2003, 254(2):261~272. doi: 10.1016/S0926-860X(03)00488-5
-
[17]
Z Nawaz. Rev. Chem. Eng., 2015, 31(5):413~436.
-
[18]
B V Vora. Top. Catal., 2012, 55(19~20):1297~1308. doi: 10.1007/s11244-012-9917-9
-
[19]
Y Nagai, T Hirabayashi, K Dohmae et al. J. Catal., 2006, 242(1):103~109. doi: 10.1016/j.jcat.2006.06.002
-
[20]
L W Lin, T Zhang, J L Zang et al. Appl. Catal., 1990, 67(1):11~23. doi: 10.1016/S0166-9834(00)84428-0
-
[21]
R D Cortright, J A Dumesic. J. Catal., 1995, 157(2):576~583. doi: 10.1006/jcat.1995.1322
-
[22]
B M Nagaraja, C H Shin, K D Jung. Appl. Catal. A, 2013, 467:211~223. doi: 10.1016/j.apcata.2013.07.022
-
[23]
C L Yu, H Y Xu, Q J Ge et al. J. Mol. Catal. A, 2007, 266(1~2):80~87. doi: 10.1016/j.molcata.2006.10.025
-
[24]
M H Lee, B M Nagaraja,K Y Lee et al. Catal. Today, 2014, 232(4):53~62. doi: 10.1016/j.cattod.2013.10.011
-
[25]
A Virnovskaia, S Morandi, E Rytter et al. J. Phys. Chem. C, 2007, 111(40):14732~14742. doi: 10.1021/jp074686u
-
[26]
B K Vu, M B Song, I Y Ahn et al. Catal. Today, 2011, 164(1):214~220. doi: 10.1016/j.cattod.2010.10.007
-
[27]
G J Siri, G R Bertolini, M L Caselle et al. Mater. Lett., 2005, 59(18):2319~2324. doi: 10.1016/j.matlet.2005.03.013
-
[28]
B M Nagaraja, H Jung, D R Yang et al. Catal. Today, 2014, 232:40~52. doi: 10.1016/j.cattod.2013.10.070
-
[29]
L Y Bai, Y M Zhou, T W Zhang et al. Catal. Lett., 2009, 129(3):449~456. doi: 10.1007/s10562-008-9822-9
-
[30]
J Silvestre-Albero, J C Serrano-Ruiz, A Sepvlveda-Escribano et al. Appl. Catal. A, 2005, 292(1):244~251. doi: 10.1016/j.apcata.2005.06.005
-
[31]
G Siddiqi, P Sun, V Gavita et al. J. Catal., 2010, 274(2):200~206. doi: 10.1016/j.jcat.2010.06.016
-
[32]
P Sun, S Georges, M Chi et al. J. Catal., 2010, 274(2):192~199. doi: 10.1016/j.jcat.2010.06.017
-
[33]
A D Ballarini, P Zgolicz, I M J Vilella et al. Appl. Catal. A, 2010, 381(1~2):83~91. doi: 10.1016/j.apcata.2010.03.053
-
[34]
S A Bocanegra, A A Castro, O A Scelza et al. Appl. Catal. A, 2007, 333(1):49~56. doi: 10.1016/j.apcata.2007.09.002
-
[35]
H Seo, K Lee, G Hong et al. J. Nanosci. Nanotech., 2015, 15(10):8318~8323. doi: 10.1166/jnn.2015.11243
-
[36]
M Setnicka, Z Tisler, D Kubicka et al. Top. Catal., 2015, 58(14):866~876.
-
[37]
R D Cortright, J M Hill, J A Dumesic. Catal. Today, 2000, 55(3):213~223. doi: 10.1016/S0920-5861(99)00249-7
-
[38]
K G Azzam, G Jacobs, W D Shafer et al. Appl. Catal. A, 2010, 390(1~2):264~270. doi: 10.1016/j.apcata.2010.10.022
-
[39]
C Jaye, M Tilyard. Fuel Proc. Technol., 2007, 88(9):883~889. doi: 10.1016/j.fuproc.2007.04.007
-
[40]
B K Vu, M B Song, I Y Ahn et al. Appl. Catal. A, 2011, 400(1~2):25~33. doi: 10.1016/j.apcata.2011.03.057
-
[41]
V Galvita, G Siddiqi, P Sun et al. J. Catal., 2010, 271(2):209~219. doi: 10.1016/j.jcat.2010.01.016
-
[42]
P L D Cola, R Glaser, J Weitkamp. Appl. Catal. A, 2006, 306(143):85~97.
-
[43]
Y Zhang, Y Zhou, J Shi et al. J. Mol. Catal. A, 2014, 381(1):138~147. doi: 10.1016/j.molcata.2013.10.007
-
[44]
S J Zhou, Y M Zhou, Y W Zhang et al. J. Mol. Catal. Sci., 2014, 49(3):1170~1178.
-
[45]
S Delsarte, F Mauge, P Grange. J. Catal., 2001, 202(1):1~13. doi: 10.1006/jcat.2001.3270
-
[46]
S Bocanegra, A Ballarini, P Zgolicz et al. Catal. Today, 2009, 143(3):334~340.
-
[47]
Z Nawaz, F Baksh, J Zhu et al. J. Ind. Eng. Chem., 2013, 19(2):540~546. doi: 10.1016/j.jiec.2012.09.024
-
[48]
J C Serrano-Ruiz, A Sepulveda-Escribano, F Rodriguez-Reinoso. J. Catal., 2007, 246(1):158~165. doi: 10.1016/j.jcat.2006.12.004
-
[49]
M Ohta, Y Ikeda, A Igarashi. Appl. Catal. A, 2004, 266(2):229~233. doi: 10.1016/j.apcata.2004.02.011
-
[50]
B Gu, S He, X Rong et al. Catal. Lett., 2016, 146(8):1415~1422. doi: 10.1007/s10562-016-1767-9
-
[51]
A H Dong, K Wang, S Z Zhu et al. Fuel Proc. Technol., 2017, 158:218~225. doi: 10.1016/j.fuproc.2017.01.004
-
[52]
R Grabowski, B Grzybowska, J Slczynski et al. Appl. Catal. A, 1996, 144(1~2):335~341. doi: 10.1016/0926-860X(96)00117-2
-
[53]
M Hoang, J F Mathews, K C Pratt. J. Catal., 1997, 171(1):320~324. doi: 10.1006/jcat.1997.1799
-
[54]
A Hakuli, A Kytokivi, A O I Krause et al. J. Catal., 1996, 161(1):393~400. doi: 10.1006/jcat.1996.0197
-
[55]
E Rombi, M G Cutrufello, V Solinas et al. Appl. Catal. A, 2003, 251(2):255~266. doi: 10.1016/S0926-860X(03)00308-9
-
[56]
V Z Vladimir, R Fridman, M Severance. Appl. Catal. A, 2016, 523:39~53. doi: 10.1016/j.apcata.2016.05.008
-
[57]
B M Weckhuysen, R A Schoonheydt. Catal. Today, 1999, 51(2):223~232. doi: 10.1016/S0920-5861(99)00047-4
-
[58]
D Sanfilippo, I Miracca. Catal. Today, 2006, 111(1~2):133~139. doi: 10.1016/j.cattod.2005.10.012
-
[59]
D R Fang, J B Zhao, W J Li et al. J. Energy Chem., 2015, 24(1):101~107. doi: 10.1016/S2095-4956(15)60290-X
-
[60]
R L Puurunen, B M Weckhuysen, J. Catal., 2002, 210(2):418~430. doi: 10.1006/jcat.2002.3686
-
[61]
B M Weckhuysen, L M D Ridder, P J Grobet et al. J. Phys. Chem., 1995, 99(1):320~326. doi: 10.1021/j100001a048
-
[62]
P P Li, W Z Lang, K Xia et al. Appl. Catal. A, 2016, 522:172~179. doi: 10.1016/j.apcata.2016.05.007
-
[63]
S D Rossi, G Ferraris, S Fremitti et al. Appl. Catal. A, 1993, 106(1):125~141. doi: 10.1016/0926-860X(93)80160-R
-
[64]
S T Korhonen, S M K Airaksinen, M A Banares et al. Appl. Catal. A, 2007, 333(1):30~41. doi: 10.1016/j.apcata.2007.08.040
-
[65]
S Udomsak, R G Anthony. Ind. Eng. Chem. Res., 1996, 35(1):47~53. doi: 10.1021/ie950205u
-
[66]
S Kilicarslan, M Dogan, T Dogu. Ind. Eng. Chem. Res., 2013, 52(10):3674~3682.
-
[67]
T A Bugrova, N N Litvyakova, G V Mamontov. Kinet. Catal., 2015, 56(6):758~763. doi: 10.1134/S0023158415060014
-
[68]
T Otroshchenko, J Radnik, M Schneider et al. Chem. Commun., 2016, 52(52):8164~8167. doi: 10.1039/C6CC02813F
-
[69]
蒋晓阳, 周广林, 樊彤彤等. 化工进展, 2016, 35(1):150~156. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hgjz201601027&dbname=CJFD&dbcode=CJFQ
-
[70]
B Zheng, W Hua, Y Yue et al. J. Catal., 2005, 232(1):143~151. doi: 10.1016/j.jcat.2005.03.001
-
[71]
N S Nesterenko, O A Ponomoreva, V V Yuschenko et al. Appl. Catal. A, 2003, 254(2):261~272. doi: 10.1016/S0926-860X(03)00488-5
-
[72]
G Wang, C Li, H Shan. Catal. Sci. Technol., 2016, 6(9):3128~3136. doi: 10.1039/C5CY01950H
-
[73]
J Han, G Jiang, S Han et al. Catalysts, 2016, 6(1):13. doi: 10.3390/catal6010013
-
[74]
G Wang, Z Meng, J Liu et al. ACS Catal., 2013, 3(12):2992~3001. doi: 10.1021/cs400705p
-
[75]
Y N Sun, Y N Gao, Y Wu et al. Catal. Commun., 2015, 60:42~45. doi: 10.1016/j.catcom.2014.11.024
-
[76]
G Wang, C Li, H Shan. ACS Catal., 2014, 4(4):1139~1143. doi: 10.1021/cs5000944
-
[77]
G Wang, C Gao, X Zhu et al. ChemCatChem, 2014, 6(8):2305~2314. doi: 10.1002/cctc.201402173
-
[78]
Y N Sun, L Tao, T You et al. Chem. Eng. J., 2014, 244(10):145~151. doi: 10.1016/j.cej.2014.01.047
-
[79]
G Wang, C Li, H Shan et al. Ind. Eng. Chem. Res., 2013, 52(27):13297~13304.
-
[80]
Y Li, J Zhang, J Wang et al. Chin. J. Catal., 2015, 36(8):1214~1222. doi: 10.1016/S1872-2067(15)60914-7
-
[81]
G Wang, X Zhu, J Zhang et al. RSC Adv., 2014, 4(100):57071~57082. doi: 10.1039/C4RA08849B
-
[82]
许燕丽, 王希涛. 化学工业与工程, 2016, 33(2):17~22. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxgy201602003&dbname=CJFD&dbcode=CJFQ
-
[83]
Y L Xu, H X Sang, K Wang et al. Appl. Surf. Sci., 2014, 316:163~170. doi: 10.1016/j.apsusc.2014.07.119
-
[84]
Y L Xu, X T Wang, L Rong. React. Kinet. Mech. Catal., 2014, 113(2):393~406. doi: 10.1007/s11144-014-0738-7
-
[85]
M A Chaar, D Patel, M C Kung et al. J. Catal., 1987, 105(2):483~498. doi: 10.1016/0021-9517(87)90076-5
-
[86]
Y P Tian, P Bai, S M Liu et al. Fuel Proc. Technol., 2016, 151:31~39. doi: 10.1016/j.fuproc.2016.05.024
-
[87]
U Rodemerck, S Sokolov, M Stoyanova et al. J. Catal., 2016, 338:174~183. doi: 10.1016/j.jcat.2016.03.003
-
[88]
Z Zhang, Y Li, J Wang et al. Catal. Sci. Technol., 2016, 6:4863~4871. doi: 10.1039/C5CY02058A
-
[1]
-

图 1 炼化企业C4烃加工利用现状
Figure 1 Current situation of exploitation of C4 hydrocarbons from refinery enterprises


表 1 异丁烷直接脱氢制异丁烯催化剂汇总
Table 1. Summary of catalysts for isobutane direct dehydrogenation to isobutene
类别 特点 举例 目前研究方向 PtSn基负载型催化剂 易活化C-H键,脱氢活性高,可以通过控制活性Pt物种尺寸以抑制副反应进行,但是易结焦且成本高 Pt-Sn-K/γ-Al2O3[14] 助催化剂、载体的选择以及制备,力求降低成本的同时提高催化剂的活性、稳定性和寿命 CrOx基催化剂 适用于直接脱氢和氧化脱氢,成本低,但是失活迅速,在高温下不稳定,且高价Cr对人体健康和环境保护不利 Cr2O3-K2O/Al2O3[15] 载体、助催化剂的选择以及催化脱氢机理的研究 GaOx基催化剂 适用于直接脱氢和氧化脱氢,含有大量的弱Lewis酸中心,催化剂上稳定存在的β-Ga2O3是脱氢活性中心 Ga2O3/MTS,Ga/Si=0.05[16] 包括载体和助催化剂的研究 金属硫化物催化剂 活性和选择性高,初始收率高于商业化的CrOx和Pt-Sn催化剂,但是几小时后催化剂因硫以H2S形式流失而迅速失活,对装置材料的抗腐蚀性要求较高 CoS/SiO2、FeS/SiO2[68] 提高催化剂的寿命和稳定性的研究 其他 — Mo/MgAl2O4、碳基材料、Co-、Ni2P-、VOx基催化剂等 研究具有高脱氢活性、选择性以及稳定性的新型脱氢催化剂  下载: 导出CSV
下载: 导出CSV
表 2 Pt基催化剂催化性能参数汇总
Table 2. Summary of the catalytic data of the Pt-based catalysts
催化剂 制备方法 反应温度/℃ 空速 进料组成 转化率/% 选择性/% 文献 2(wt)% Pt-1(wt)%
Sn/CeO2/C共浸渍 500 WHSV=6h-1 H2/i-C4H10=6 37~20 90~100 [48] Pt-Sn/MA-P1 真空等体积浸渍 580 1200mL i-C4H10/gCat. H2/i-C4H10=1:1 55.8~53.7b 65b [50] PtSn-La/Al2O3 分步浸渍 590 10mL i-C4H10/gCat. H2=10mL/min
i-C4H10=5mL/min49.4c 97c [51] PtSnK/γ-Al2O3d 分步浸渍 590 GHSV=3000h-1 H2/i-C4H10=2 29.0 94 [14] Pt-Sn/slit-SAPO-34e 共浸渍 585 WHSV=5h-1 H2/i-C4H10=0.2 40.1~16.0 84 [47] a0.2(wt)% Pt,且Sn/Pt=0.5,并标明了初始化转化率及选择性;b分别为始末转化率和初始选择性;c分别为反应220min时刻的转化率和选择性;d催化剂配比为0.5(wt)% Pt,0.5(wt)% Sn,0.3(wt)%K,且标明了反应6h后的转化率和选择性;e催化剂配比为0.5(wt)% Pt,1(wt)% Sn,并标明了初始转化率及反应8h后的选择性。
下载: 导出CSV
表 3 CrOx基催化剂催化性能参数汇总
Table 3. Summary of the catalytic data of the CrOx-based catalysts
催化剂 制备方法 反应温度/℃ 空速 原料组成 转化率/% 选择性/% 文献 3(wt)% Cr-MCM-41 原位水热合成 600 WHSV=26 h-1 i-C4H10=20 mL/min 27a - [66] 15(wt)% Cr-Al2O3b 浸渍 600±5 - 纯i-C4H10 63~30 90 [59] CrOx/nZrO2-SiO2c 等体积浸渍 540 GHSV=540 h-1 i-C4H10=270 mL/min
N2=1530 mL/min- >96 [67] Cr10Zr90Od 共浸渍 550 0.24 g·s·mL-1 i-C4H10/H2=40:60 36 84 [68] K2O-Cr2O3-B2O3/γ-Al2O3e 等体积浸渍 560 400 h-1 H2/i-C4H10=6 57.1~36.4 92.6~93.2 [69] a反应15min时达到最大值;b标明了其始末转化率及反应750min时的选择性;c n=1.0~2.0 (wt)%,催化活性为反应15min时的选择性;d标明了反应9min时的初始转化率及选择性;e2 (wt)% K2O,12 (wt)% Cr2O3,1 (wt)% B2O3,并分别标明了反应0.5h和8h时的转化率和选择性。
下载: 导出CSV
表 4 讨论的其他类型脱氢催化剂催化性能参数汇总
Table 4. Summary of the catalystic data of other types dehydrogenation catalysts discussed
催化剂 制备方法 反应温度/℃ 空速 原料 转化率/% 选择性/% 收率/% 反应时间/h 文献 Mo/MgAl2O3 等体积浸渍 560 GHSV=54.5 h-1 i-C4H10=2mL/min
N2=12mL/min78a 45a 35a 200 [79] CSACb - 625 WHSV=6000mL·g-1·h-1 i-C4H10=1mL/min
N2=19mL/min70 76 53.2 1 [80] OMC8c 浸渍 620 6000mL·g-1·h-1 i-C4H10=1mL/min
N2=19mL/min38 81 30.8 66.7 [88] 13(wt)% Co/SiO2d 等体积浸渍 560 - i-C4H10=2mL/min
N2=12mL/min71e 97e 61.8e 2 [81] Ni2P/SiO2 浸渍 460 800h-1 i-C4H10=2mL/min
N2=12mL/min~4.6e ~72e 80e 2 [82] 6.5(wt)% Cs-Ni2P/SiO2 等体积浸渍 460 1600h-1 i-C4H10/N2=10/10 6 96 5.8 4 [84] Ni2P/AC 等体积浸渍 560 1200h-1 i-C4H10/N2=1/1 15 89 13.4 4.5 [83] 10(wt)%VOx-K2O/γ-Al2O3 等体积浸渍 590 2250mL·g-1·h-1 i-C4H10/N2=1/1 ~61 ~70 42.5 1.75 [86] 注:a催化剂装填量为4g(80~180 μm),数值为平均值;bCSAC为椰壳形活性炭;COMC8(有序介孔碳)经800℃焙烧2h;d经硫化处理2h;e为初始活性。
下载: 导出CSV
-








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 2141
- HTML全文浏览量: 389
 下载:
下载:
