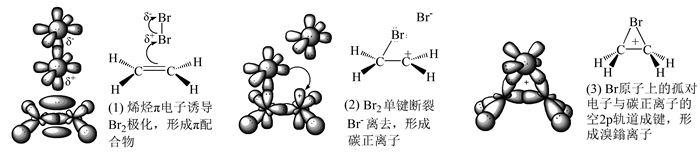
图 1.
经典的溴鎓离子的形成过程示意图
Figure 1.
Classical diagrammatic sketch of the formation of the bromonium ion

烯烃与溴的加成通常具有高度立体专一性,因而在有机化学的立体合成领域一直备受关注。早在1937年,Bartlett和Tarbell就已经提出该加成反应的中间体是环状桥联溴鎓离子的假说,但是该离子直到30年后才于强酸性介质中获得,并借助当时开拓性的核磁共振研究完成结构的鉴定。该假说提出后不久即被广泛接受,因为它几近完美地解释了反式加成的原因[1]。目前,教材一致采用了环溴鎓离子中间体来解释烯烃与溴的加成历程,较常见的溴鎓离子形成过程如图 1所示[2, 3]。然而,教材对溴鎓离子的精细结构一直缺乏明确的阐释,尤其是三元环中各原子的杂化方式。而且,教材普遍接受的溴鎓离子为σ-配合物的说法在研究性文献中并无定论。此外,已有的文献和网络资源对三元环上的电荷分布情况并没有一致的观点,这严重影响了对Br-亲核进攻过程的透彻理解。鉴于以上原因,笔者在查阅了国内外大量文献的基础上,将重点对以上诸问题进行解析,以期为教材中溴鎓离子的知识介绍提供一定的补充。
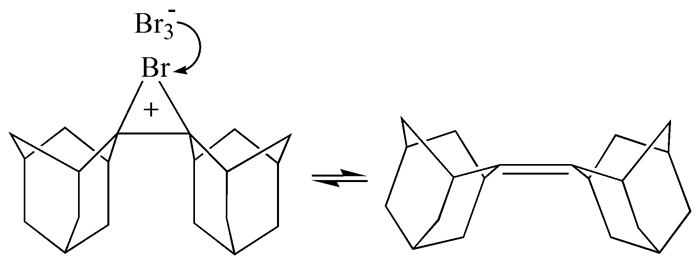
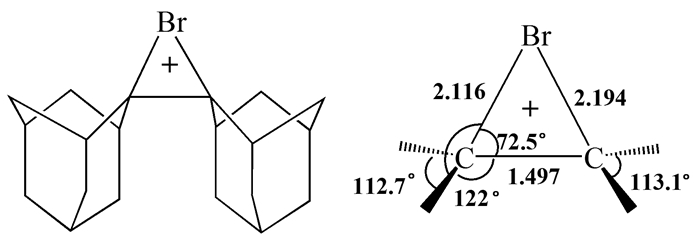
自溴鎓离子环状桥联结构假说提出以后,直到1969年Strating等才首次发现非质子溶剂中因亲核试剂的进攻受到空间位阻的抑制,溴与金刚烷烯的加成会停留在溴鎓离子的形成这一步,从而确立了金刚烷烯溴鎓离子的制备方法。该离子的一个重要反常特征是它的不对称性,两个C-Br键长相差0.078Å。然而,这种不对称性可能源于晶体堆积中三溴化物反离子的限制,在溶液中未必如此[4]。迄今为止,金刚烷烯溴鎓离子(图 2)仍是唯一稳定可分离并且其晶体结构被检测的溴鎓离子中间体,故此在很多情况下其他溴鎓离子的结构是从该离子的结构推测出来的。
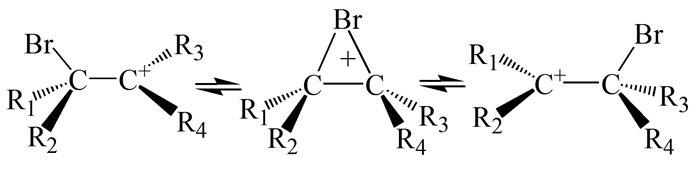
Olah等[5]曾在亲核超强酸介质中获得了甲基取代的乙烯溴鎓离子的1H和13C NMR谱,并且从对称或不对称桥联溴鎓离子与开链β-溴碳正离子之间平衡的角度讨论了三元环碳原子的化学位移与甲基数目的关系。偕-二取代基在桥联结构中制造的不对称会有利于图 3所示平衡关系的建立。另一碳原子上第三个甲基的取代将会使这种建立平衡关系的优势更加突出。据此,Olah等推测溴鎓离子结构可能是开链β-溴碳正离子对与桥联环状结构的混合物。然而这却未得到其他研究的证实,甚至与Galland等[6]的MNDO(改进的忽略双原子微分重叠)计算结果产生了很大的矛盾(第3节中有详述),因此上述的平衡关系并不能描述乙烯溴鎓离子的结构。
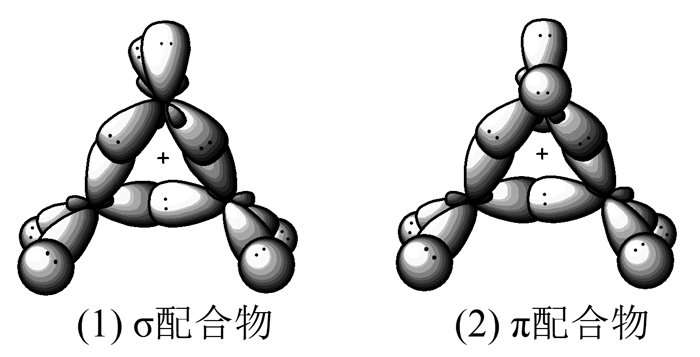
关于溴鎓离子该被视为σ还是π-配合物的问题也是理论化学家们对溴鎓离子的研究中首先关注的问题之一。问题引起争议的原因关键在于没有明确的实验标准来区分这两种键合。在早期研究中,将电荷转移复合物及溴鎓离子分别定义为π-配合物和σ-配合物,因而教材中也就沿用这种习惯命名,虽然这从轨道键合的角度来看并不严格,但一直以来还没有改进的方法。
理论计算和实验所得的键长和键角值总是介于两种配合物之间。金刚烷烯溴鎓离子的X-射线结构分析数据倾向于将其归类为σ-配合物,而乙烯溴鎓离子的从头算数据似乎又显示了其具有明显的π-配合物特征。C2H4Br+中C-Br键长经从头算量子力学技术计算为0.2025nm,这比溴代烃中典型的C-Br键(0.194nm)长了少许;而经X-射线检测金刚烷衍生物三元环中C-Br键长比理论计算值又增加了0.13nm。造成该差异的原因可能是金刚烷衍生物使乙烯的平面变形少,导致中间体更像π-配合物,从而弱化了C-Br,键长增长。C2H4Br+中C-C键长为0.1455nm,介于典型的C—C(0.154nm)和C=C(0.134nm)之间。金刚烷取代物C-C键长0.1497nm,这对π-配合物来说几乎达到极限,因为这恰好等于环丙烷的C-C键长。有研究者认为这可以通过空间位阻来解释,但是未溴化的金刚烷烯的C-C键长为0.1336nm。溴化后0.0161nm的增长看起来更像σ-配合物。因此,X-射线技术所鉴定的结构对认清溴鎓离子三元环状结构的本质几乎毫无作用。而C2H4Br+从头算计算的C-C键长(0.1455nm)也只是略高于Itell等[7]所给出的π-配合物C-C(0.134~0.144nm)的上限。CH2距离先前所在的C2H4平面大约向下倾斜17.4°,介于乙烯和环丙烷(30°)之间,而π-配合物中CH2对平面的典型变形恰为16°~20°。
综上所述,计算和实验所得的结果都未能得到任何确凿的结论,正像Slebocka-Tilk等[8]总结的那样,关于溴鎓离子是σ还是π-配合物的区分仅仅停留在字面上而已,已经没有任何实质化学意义。
溴鎓离子的结构扑朔迷离,三元环上各原子的杂化方式更是难以言明,而关于环上的电荷分布在诸多文献和教材上也很难找到确定的描述。虽然Br-与溴鎓离子中C的亲核反应为SN2历程已成为公认的事实[2],但据此判断三元环上的正电荷分布情况却并不严谨。从整个反应历程来看,Br-的亲核反应生成最终产物的过程比前一步溴鎓离子的生成过程快了许多,表面上看正电荷似乎只驻留在碳原子上,这在最初还曾让人们误以为溴鎓离子的生成为不可逆过程。Strating等[4]研究证实,在高稳定的金刚烷烯溴鎓离子与Br3-共存的体系中有金刚烷烯释放出来(图 4)。后来,Gedye等证实在Br-存在时,环己烯溴鎓离子也可裂解出游离溴[9],但其高温、高溴化物浓度和反应时间长等极端的反应条件令人对该发现的普遍性深表质疑。这些实验事实说明,三元环上的电荷并不单单只驻留在某一原子上。
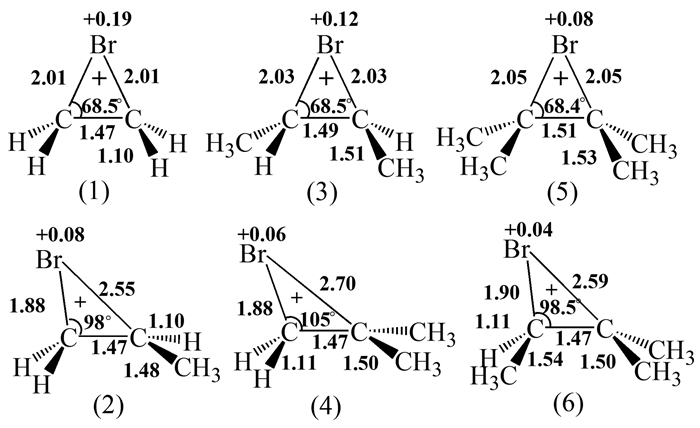
Galland等[6]经从头算法得到若干种甲基取代溴鎓离子的结构,包括某些重要的键长、键角和电荷分布情况等信息(图 5)。对称取代的溴鎓离子(1)、(3)、(5)具有等角度的精确对称结构,且与甲基的数目无关。三种溴鎓离子的θBr-C-C和Br上的电荷都呈下降趋势,这说明取代烷基增加了碳原子的阳离子特性。不对称取代的溴鎓离子(2)、(4)、(6)的结构高度不对称。取代基最少的碳原子接近sp3杂化(准sp3杂化),而取代基最多的碳原子接近于sp2杂化。然而,即使是(4)和(6)这样的离子也不能被完全视为β-溴碳正离子,这是因为θBr-C-C和C-C键并没有像预想那样彻底失去Br-C(sp2)相互作用影响。它们的数值与2-溴丙烷之间相对应的量值(dC-Br=0.1944nm,θBr-C-C=116.2°)还有一定距离。
由此可见,对乙烯溴鎓离子来说,如若非要从杂化轨道理论的角度分析其三元桥联结构的话,则有必要借鉴其他已知三元环结构来寻找可能的答案,譬如含σ键的环丙烷(图 6(1)),环氧乙烷(图 6(2))以及含π键的环丙烯碳正离子(图 6(3))等。
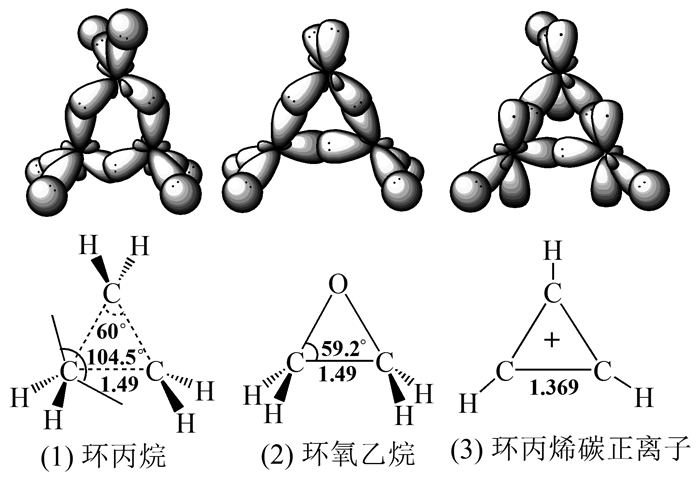
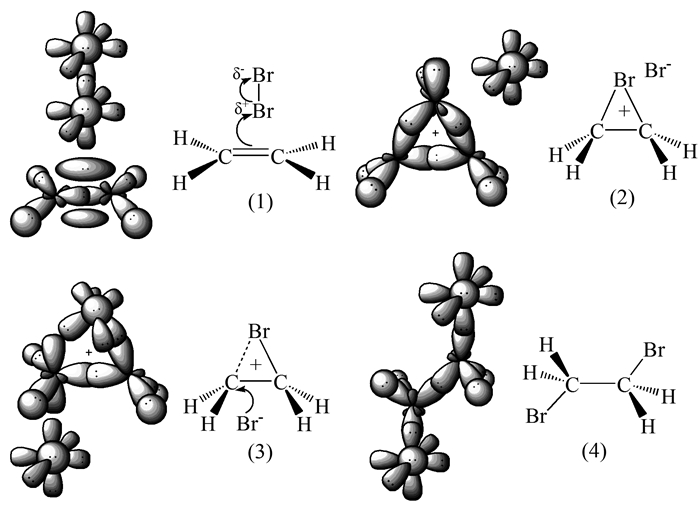
若乙烯溴鎓离子为σ-配合物,则其三元环结构类似于环丙烷和环氧乙烷。教材中以往经典的论述认为在溴鎓离子的形成过程中溴4p轨道上的孤对电子与中间态碳正离子未杂化的空2p轨道成键,即认为成键过程中Br原子未发生杂化。然而,4p和4s轨道上的另两对孤对电子虽能实现环平面两侧的对称,但却与已成键的另两个4p轨道之间的距离较近,从能量角度讲,稳定性会较差。因此推测,Br-离去后,与乙烯作用的Br应像环丙烷中的C及环氧乙烷中的O一样发生sp3杂化,两个单电子的sp3杂化轨道与C的准sp3杂化轨道成键(Br-离去后C杂化方式亦发生改变,接近sp3杂化),余下的由两对孤对电子占据的sp3杂化轨道就会以稳定的对称位置分踞于环平面的两侧,增大了溴鎓离子的稳定性(图 7(1))。相反,若乙烯溴鎓离子为强π-配合物,则其三元结构类似于环丙烯碳正离子。乙烯诱导Br2极化后,π电子与带部分正电荷的Br原子作用加强,导致Br-Br键断裂后,与烯烃作用的Br原子随即变成sp2杂化(图 7(2)),两个单电子分占两个sp2杂化轨道,与C的准sp3杂化轨道成键(Br-离去后C杂化方式亦发生改变),而未杂化的4p轨道上有一对孤对电子,可与C的4个C-H键发生部分重叠,类似于超共轭,这是由于C的杂化方式介于sp2和sp3之间,C-H键距离Br的4p轨道较近。故此,计算化学的数据显示溴鎓离子表现出π-配合物的某些特征就很容易理解。
综上所述,溴鎓离子无论属于哪种配合物,其三元环上的Br原子都应以杂化方式成键。教材中常见的溴鎓离子生成历程的第二步所出现的碳正离子,其实已经引起人们的质疑[10]。尽管它没有三元溴鎓离子稳定,但是如若在反应历程中出现,就势必会给Br-造成进攻机会,那么终产物中就一定有顺式产物出现,而实验事实恰恰相反。长期以来,人们只关注三元环上C的杂化方式,而忽略Br的杂化。对比其他已知的三元环结构可知,溴鎓离子中Br的杂化方式可能与C一样介于sp3和sp2杂化之间,当溶剂和取代基等因素对其产生影响时,三元环上的各原子则趋向于采用产生最稳定结构的杂化方式。
在此,对乙烯与Br2的加成历程作以修正和总结(图 8,以乙烯溴鎓离子为σ-配合物为例,π-配合物的情况与此类似,不再赘述):(1)烯烃π电子诱导Br2发生极化,形成配合物;(2)Br-Br键断裂,Br-离去,余下的Br原子随即发生sp3杂化,与C形成三元环,由于C和Br的电负性差异,导致正电荷主要驻留在C原子上,Br上亦有少量正电荷,因此C成为Br-下一步亲核进攻三元环的主要目标,但Br也有接受进攻的机会;(3)Br-亲核进攻C后,三元环上的C-Br键变弱,C趋向于sp2杂化,Br趋向于不杂化;(4)随Br-与C的作用进一步加强,C变典型的sp3杂化,新的C-Br键形成,环上C-Br键断裂,Br作为离去基团也形成另一新的C—Br键。
虽然溴鎓离子的三元环具有诸多特殊性,其结构至今也无明确的结论,但各教材上简要的介绍,还是存在某些矛盾以及不足之处。因此,笔者从教学视角出发,概述了溴鎓离子结构的研究成果,并以教学中常用的杂化轨道理论探讨了溴鎓离子三元环上各个原子可能的杂化方式。基于计算和实验研究所得的有关溴鎓离子结构的结论至今仍存在分歧,但根据这些结果可以推断三元环上的C、Br原子皆非地道的sp2或sp3杂化,而是处于二者之间,更倾向于哪种杂化取决于两个C上的取代基或溶剂的种类等因素,因为它们会对溴鎓离子的稳定性存在至关重要的影响。
T P Hamilton, H F Schaefer. J. Am. Chem. Soc., 1990, 112(23):8260~8265. doi: 10.1021/ja00179a006
裴伟伟. 有机化学核心教程. 北京:科学出版社, 2008.
M Jones, S A Fleming. Organic Chemistry. New York:WW Norton & Company, Inc. 2010.
J Strating, J H Wieringa, H Wynberg. Chem. Commun., 1969, 16:907~908.
G A Olah, P W Westerman, E G Melby et al. Chem. Commun., 1974:96(11):3565~3573.
B Galland, E M Evleth, M F Ruasse. Chem. Commun., 1990, 13:898~900.
S D Ittel, J A Ibers. Adv. Organomet. Chem., 1976, 14:33~61. doi: 10.1016/S0065-3055(08)60648-6
H Slebocka-Tilk, R G Ball, R S Brown. J. Am. Soc. Chem., 1985,107(15):450~4508.
R Gedye, R S Brown, H Slebocka-Tilk et al. J. Am. Soc. Chem.,1984, 106(16):4515~4521. doi: 10.1021/ja00328a035
李晓丽, 许苗军, 孙才英 等. 大学教育, 2012, 1(4):60~61. http://www.cnki.com.cn/Article/CJFDTotal-DXJY201204030.htm

图 1 经典的溴鎓离子的形成过程示意图
Figure 1 Classical diagrammatic sketch of the formation of the bromonium ion

图 3 溴鎓离子与开链β-溴碳正离子对之间的平衡
Figure 3 Equilibrium between the bromonium ions and a pair of open β-bromocarbocations

图 4 金刚烷烯溴鎓离子的可逆生成过程
Figure 4 Reversible formation of the adamantylideneadamantane bromonium ion

图 5 几种甲基取代溴鎓离子重要键长、键角及Br上的电荷分布
Figure 5 Some critical bond distances and angles of several methyl-substituted bromonium ions as well as the charge on the Br atom

图 6 几种三元环中原子的杂化及结构图
Figure 6 Diagrams of structure and hybridization of atoms in several three membered rings

图 7 溴鎓离子中各原子可能的杂化方式
Figure 7 Possible hybridization modes of various atoms in the bromonium ion

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

