图 1.
改性前后催化剂的脱硝活性
Figure 1.
SCR activity of the unmodified and modified catalysts

氮氧化物(NOx)是大气污染物的有害物质之一, SCR是非常有效的脱除氮氧化物的过程, 工业催化剂为WO3-V2O5/TiO2催化剂, 通常适用温度在300-400 ℃, 活性温度窗口窄[1], 并且含有有毒的钒。近年来,众多研究者致力于开发新的环保型催化剂。
锰氧化物具有多种价态, 有利于氧化还原反应的进行, 是目前研究较多的NH3-SCR催化剂。工业应用中, 通常将活性组分负载在不同的载体上, 如分子筛、二氧化钛或氧化铝。载体不仅提供机械强度, 其比表面积和酸性位也是影响脱硝活性的重要参数。Kang等[2]对负载型锰基SCR催化剂的载体进行了系统的研究, 催化剂活性按照MnOx/TiO2≈MnOx/γ-Al2O3>MnOx/SiO2>MnOx/Y-ZrO2的顺序递减。说明氧化铝与二氧化钛活性接近, 但其比表面积更大。但Mousavi等[3]认为, Mn/γ-Al2O3催化剂在269.8 ℃时活性最高, NOx脱除率可达94.6%, 活性温度较高。而郭静等[4]制备了Mn-Ce/Al2O3双活性组分催化剂, 在200 ℃达到90%以上, 铈的引入显著降低了Mn/Al2O3的脱硝温度。Jin等[5]对比研究了Mn-Ce/TiO2和Mn-Ce/Al2O3催化剂, 发现当温度高于150 ℃时, 以Al2O3为载体的催化剂具有更高的活性, 证明氧化铝上适当的B酸有利于提高脱硝活性, 提高脱硝活性温度。
烟气是一种复杂的混合物, 除了氮氧化物外, 还存在硫化物和水蒸气, 一般认为, 二氧化硫会毒化催化剂, 降低催化剂的脱硝活性[6, 7]。但也有研究者[8, 9]认为, 原料气中二氧化硫会增加表面活性氧, 改变氧化能力;形成的硫酸盐能增强表面B酸和L酸浓度, 改变对氨的吸附, 增加脱硝活性。对于活性金属(氧化铈)硫化前后的脱硝性能的影响已有研究报道[10], 发现氧化铈可以形成硫酸盐并能提供脱硝活性, 但形成体相硫酸盐后, 由于活性位的缺少, 会影响脱硝活性[11]。因此, 关于脱硝催化剂上硫酸盐对脱硝活性的影响还需要进一步深入研究。此外, 大部分学者仅研究了活性金属硫酸化或成型催化剂硫酸化处理的情况, 这会减少活性组分的数量。而单独对载体进行硫酸化处理, 再负载金属可以避免活性组分数量的减少, 这种改性方式对脱硝性能的影响还极少有详细的研究报道。
本研究先采用不同浓度的硫酸溶液对氧化铝载体进行预处理改性, 制备不同酸性的改性氧化铝, 然后再在改性氧化铝载体上浸渍相同含量的锰和铈, 研究氧化铝载体预处理对催化剂性质及脱硝活性的影响。
氧化铝(Al2O3), AR, 国药集团化学试剂有限公司;硝酸锰溶液(Mn(NO3)3, 50%(质量分数) in H2O), AR, 国药集团化学试剂有限公司;硝酸铈·六水合物(Ce(NO3)3·6H2O), AR, 国药集团化学试剂有限公司;硫酸(H2SO4)(98%, 质量分数), AR, 国药集团化学试剂有限公司。
采用浸渍法制备, 首先, 称取一定量的80-180目的氧化铝小球载体;然后, 称取硝酸锰和硝酸铈, 并加水充分溶解, 分三次将配制好的溶液浸渍在载体上, 将每次浸渍完的催化剂在60 ℃烘箱中烘干;最后将浸渍并烘干的催化剂在500 ℃焙烧3 h。催化剂命名为M/Al2O3, 其MnO2和CeO2金属质量分数分别为5%和4%。
首先对Al2O3进行改性, 改性步骤为:配制不同浓度的硫酸溶液, 将氧化铝颗粒与酸溶液在室温下混合搅拌, 固液比为1 g/5 mL。然后进行固液分离, 在室温下缓慢干燥24 h, 600 ℃焙烧2 h。然后再按照1.2.1方法负载金属, 催化剂上含有质量分数5%的MnO2和4%的CeO2。制得改性后催化剂命名为M/xS/Al2O3,其中,x表示硫酸溶液的浓度, x分别为0.2、0.5、1.0。如M/0.2S/Al2O3, 代表改性硫酸浓度为0.2 mol/L。
采用荷兰帕纳科公司的X’ Pert PRO MPD衍射仪进行分子筛X射线(XRD)表征, Cu靶Kα射线, X射线波长为0.154874 nm, 管电压为40 kV, 管电流15 mA, 10°-75°, 扫描速率为10 (°)/min。
采用天津先权公司生产的TP-5079全自动多用吸咐仪进行酸性(NH3-TPD)和H2-TPR分析。
NH3-TPD:称量0.2 g样品放入吸附管中, 在氨气中加热预处理30 min。然后降低样品温度到50 ℃, 将氦气切换到氨气吸附30 min。吸附后切换为氦气吹扫并走基线。基线稳定后以10 ℃/min的升温速率升温到600 ℃, 获得该催化剂体系的NH3-TPD谱图。
H2-TPR:称量0.1 g的样品放入吸附管中, 实验所用的还原气体组成为10%H2和90%N2所组成的混合气体, 气体的体积流量为30 mL/min, 以10 ℃/min的升温速率程序升温至700 ℃, 获得该催化剂体系的H2-TPR谱图。
催化剂比表面积和孔结构采用美国康塔公司的Quadrasorb SI多用吸附仪测定。测定比表面积数据, 用Brunauer-Emmett-Teller(BET)等温方程计算样品的比表面积, 用BJH法计算催化剂的孔径和孔体积。
采用美国Termo NicoletCo.公司生产的NexusTM型傅里叶变换红外光谱仪(FT-IR)进行酸类型和原位透射吸附性能测试, 使用MCT检测器, 采集32次, 分辨率为4, 采集4000-400 cm-1的红外光谱谱图。原位透射实验中, 将样品研磨压片, 放入红外池, 在测试前在500 ℃采用N2吹扫1 h。当样品温度降到反应温度后记录为背景, 根据实验设计通入不同气体, 扫描获得谱图。
采用自制的固定床微反装置测试催化剂的SCR活性。每次测试催化剂量为0.6 g。在常规反应评价中反应气的组成为NOx(0.1%)、NH3(0.1%)、O2(5%), 气体总流量为200 mL/min。采用Testo 350-EPA烟气分析仪测试进出口气体中各组分的浓度。催化剂的脱硝率由公式(1)计算求得:
|
$ 脱硝率(\%)=\left(\varphi_{\mathrm{NO}_{x}, \mathrm{in}}-\varphi_{\mathrm{NO}_{x}, \mathrm{out}}\right) / \varphi_{\mathrm{NO}_{x}, \mathrm{in}} \times 100\% $ |
(1) |
催化剂的抗水性能是在250 ℃时通入10%H2O;抗水抗SO2性能数据是在250 ℃时通入10%H2O和0.07% SO2。
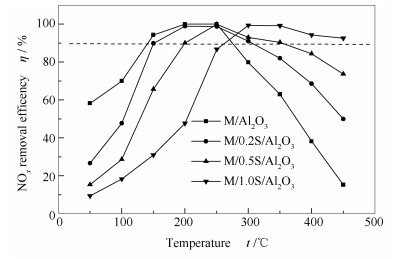
图 1为改性前后催化剂的脱硝活性。由图 1可知,随着温度的升高, 改性前后催化剂的NOx脱除率都呈现相同的变化趋势, 都是先升高后下降。但最高活性点的温度和高活性温度区间不同。M/Al2O3、M/0.2S/Al2O3、M/0.5S/Al2O3、M/1.0S/Al2O3催化剂最高活性点温度分别为200-250、200-250、250、300 ℃;大于90%活性温度范围, 依次为140-280 ℃(温度窗口为140 ℃)、150-300 ℃(温度窗口为150 ℃)、200-350 ℃(温度窗口为150 ℃)、260-450 ℃(温度窗口为190 ℃)。可见, 随着改性溶液浓度的增加, 催化剂的最高活性处的温度向高温偏移, 大于90%活性温度窗口变宽。所有催化剂脱硝活性大于90%的温度窗口大于现有工业应用的WO3-V2O5/TiO2催化剂100 ℃(300-400 ℃[1]), 并且不同催化剂适用于不同的温度范围, 可以根据实际使用的温度选择不同的改性溶液浓度。
当温度为200和250 ℃时, M/Al2O3、M/0.2S/Al2O3两催化剂的活性基本相同。当温度低于200 ℃时, 未改性催化剂的脱硝活性最高, 随着改性溶液浓度的增加, 催化剂活性不断降低。当温度高于250 ℃时, 特别是高于300 ℃以后, 未改性催化剂的脱硝活性最低, 改性溶液浓度越高, 催化剂脱硝活性越高。
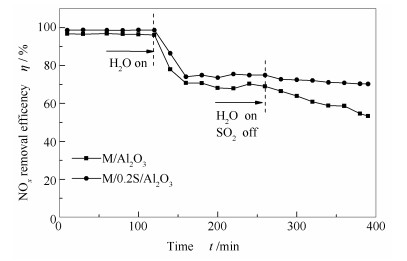
图 2为M/Al2O3和M/0.2S/Al2O3催化剂的抗水和抗硫性能。由图 1可知, 两个催化剂在250 ℃的反应活性基本相等, 因此, 选用这两个催化剂作为对比, 考察载体改性对催化剂抗水和抗硫性能的影响。由图 2可知, 在通入水后, 两个催化剂的脱硝活性都有不同程度的降低, M/0.2S/Al2O3的抗水性能略优于未改性的M/Al2O3。随后继续通入10%的H2O和0.07%的SO2, 催化剂的脱硝活性继续下降, 但M/0.2S/Al2O3具有较好的抗硫性能, 相比进水条件下, 催化剂的脱硝率略有下降, 并且在测试的时间范围内脱硝率下降缓慢, 脱硝率基本稳定在70%左右;但未改性的M/Al2O3催化剂的脱硝率一直快速下降。说明硫化物在此催化剂上一直在不断地大量生成, 并覆盖了催化剂的活性位。而载体改性可以有效抑制催化剂上硫酸盐的快速生成。
改性前后催化剂的XRD谱图见图 3。一般认为, 在28.7°、37.4°、42.9°、56.8°、59.5°的峰属于MnO2特征衍射峰[12];28.6°、33.1°、47.6°和56.4°属于CeO2的特征衍射峰[13]。33.1°和55.3°处的特征峰为Mn2O3的特征衍射峰[14]。三种氧化物有部分衍射峰相互重叠或与Al2O3重叠。由图 3可知, 在M/Al2O3催化剂中没有明显的MnO2和CeO2的特征衍射峰, 在相应的位置仅出现少量的包峰。说明金属在载体表面高度分散或是无定形的。当改性后, 催化剂中出现了不同强度的MnO2、Mn2O3或CeO2特征峰, 说明载体上预先负载的硫, 阻止了两种金属的分散, 发生了金属聚集。并且, 随着硫酸浓度的增强, 金属衍射峰强度增加, 金属颗粒变大。但并没有检测到硫与金属或氧化铝相互作用形成的新物相的衍射峰。据文献报道[15], 锰的不同氧化物对脱硝反应活性不同, 按MnO2>Mn5O8> Mn2O3> Mn3O4>MnO的次序减弱[13, 16]。无定形MnOx有利于质子的引入和释放, 从而加速反应物在催化剂体相或表面的化学吸附-脱附和氧化还原反应, 进而促进SCR反应的发生[17], 特别有利于加速低温脱硝反应活性。结合脱硝结果的差别来看, 催化剂活性相及其分散程度差别是影响催化剂脱硝活性的因素之。
改性前后催化剂的比表面积和孔结构见表 1。
 下载:
导出CSV
下载:
导出CSV
| Catalyst | Specific surface area A/(m2·g-1) | Average pore diameter d/nm | Pore volume v/(cm3·g-1) |
| Al2O3 0.2S/Al 0.5S/Al 1.0S/Al M/Al2O3 M/0.2S/Al M/0.5S/Al M/1.0S/Al |
177.3 168.1 164.3 165.2 143.2 132.1 139.1 136.3 |
3.61 3.58 3.59 3.61 3.62 4.29 3.63 3.93 |
0.24 0.25 0.25 0.25 0.21 0.21 0.22 0.21 |
由表 1可知, 与未改性的氧化铝载体相比, 改性氧化铝载体的比表面积略有降低, 孔容略微增大。这可能是改性时硫酸对氧化铝产生了侵蚀, 但并未完全破坏其骨架结构[18]。不同浓度改性的氧化铝之间比表面积差别不大, 用于后续金属浸渍具有可对比性。载体浸渍金属后, 与未改性催化剂相比, 改性后催化剂的比表面积略有降低;说明载体改性后, 影响了锰和铈的分布, 但并未造成更严重的堵孔。然而改性液浓度变化对催化剂的比表面积和孔容影响不大, 但对脱硝活性的影响却较大, 说明对于负载型催化剂, 其比表面积和孔容, 不是影响脱硝活性的关键因素。
图 4为改性前后催化剂的NH3-TPD谱图。一般认为, 脱附峰温度在100-250 ℃为弱酸, 250-400 ℃为中强酸, >400 ℃为强酸[19]。从图 4峰位置来看, 所有催化剂NH3-TPD峰温都在150-250 ℃, 属于弱酸和中强酸。但是随着改性溶液浓度的增加, 峰温逐渐升高, 表明催化剂的酸强度略有增强;同时峰面积也逐渐变大, 表明催化剂的酸性位数量不断增多。因此, 随着改性溶液浓度的增加, 催化剂的酸强度和酸量都有所增加[20]。
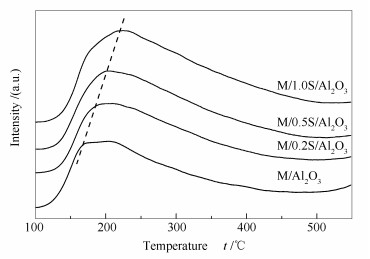
图 5为改性前后催化剂的Py-FTIR谱图。由图 5可知, 未改性的催化剂, 几乎没有B酸位, 基本全部为L酸位。经过硫酸改性后, 出现了B酸位, 并且随着硫酸浓度的增加, B酸量逐渐增加。这主要是因为表面的S元素对羟基电子云的吸引, 致使羟基中的H更容易电离产生较强的B酸[20, 21]。
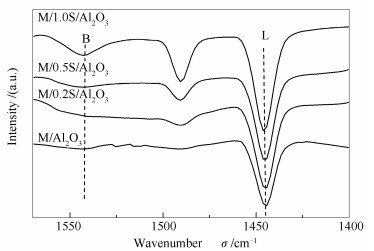
随着催化剂酸量的增加, 特别是B酸量增强, 催化剂低于200 ℃的低温脱硝活性下降, 而大于300 ℃的高温脱硝活性增强。因此, 尽管改性催化剂金属氧化物颗粒聚集, 但大于300 ℃的高温脱硝活性增强, 说明催化剂酸性的增强, 有利于增强300 ℃以上的高温脱硝活性。
催化剂氧化还原性能是影响催化剂低温催化性能的重要因素。锰的氧化物有多种形态, 其还原过程一般按照如下顺序进行:MnO2-Mn2O3-Mn3O4-MnO, 300-400 ℃的还原峰应归属于MnO2或Mn2O3→Mn3O4的还原;400-500 ℃的还原峰应归属于Mn3O4→MnO的还原[14]。
改性前后催化剂的H2-TPR谱图见图 6。在300-400 ℃, 未进行硫酸修饰的氧化铝负载金属后, 314和400 ℃两处有两个还原峰, 且400 ℃处的峰面积较小, 说明在该催化剂上可能存在两种形态的氧化锰, 也可能是高分散二氧化锰与少量聚集颗粒的二氧化锰的还原峰。由XRD谱图也发现存在很微小的MnO2的衍射峰, 还可能存在Mn2O3。当采用0.2 mol/L的硫酸溶液进行修饰时, 催化剂仅在380 ℃时存在一个峰, 接近于400 ℃, 说明与未改性的催化剂相比, 该催化剂上高分散的金属颗粒减少, 颗粒粒径略有增大, 会形成少量聚集态的二氧化锰;采用0.5和1.0 mol/L的硫酸进行修饰时, 仅剩下400 ℃附近的还原峰, 且峰面积较大, 说明此时催化剂中颗粒较大的聚集态的二氧化锰数量较多, XRD上已经明显看出晶体的衍射峰。硫酸修饰的催化剂在500-700 ℃出现新的还原峰, 540 ℃左右的还原峰应归属于氧化铈的还原峰[14]。说明改性后, 催化剂上CeO2分散度变差, 形成了较大的颗粒金属氧化物或CeO2金属与载体之间的作用力增强。并且, 改性浓度增加后, 该峰向高温偏移, 与在600 ℃硫物种[22]的分解峰重合, 最终形成了较大的还原峰。
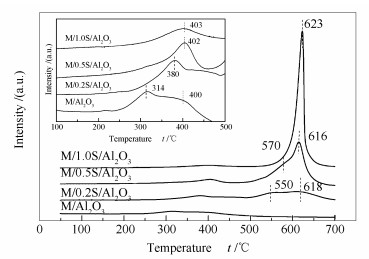
催化剂的脱硝活性与其氧化还原能力密切相关, 催化剂的低温还原峰面积越大, 表明该催化剂表面的活性氧物种数量越多, 催化剂低温活性越高[23, 16]。改性后催化剂低温还原峰面积变小, 还原峰温度变高, 说明催化剂表面可被还原的氧化物变少, 表面氧的移动能力变弱。结合XRD和BET结果可知, 改性催化剂活性组分的分散度降低, 形成了较大颗粒的晶体, 催化剂的氧化性能变差。
未改性催化剂在反应温度低于200 ℃时, 脱硝活性都较高, 与其金属颗粒高度分散及其氧化还原活性较高相对应, 说明催化剂的低温脱硝活性与其氧化还原性能密切相关。
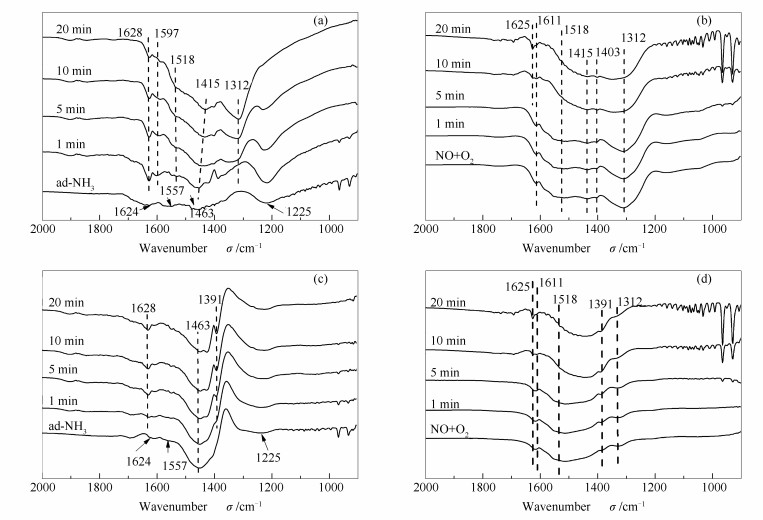
图 7为150 ℃下M/Al2O3和M/0.5S/Al2O3催化剂的FT-IR谱图。其中,图 7(a)和图 7(c)谱图中的催化剂首先在NH3中进行预吸附, 之后断开NH3, 引入不同时间的NO和O2混合气。由图 7(a)可知, M/Al2O3催化剂吸附NH3后, 在1624、1557、1463和1225 cm-1处出现吸附峰。1624和1225 cm-1归属为Lewis酸吸附NH3的非对称和对称变形振动峰[5];1557 cm-1归属为Lewis酸吸附的NH3脱氢形成的-NH2中间物种吸附峰[24, 25]。1463 cm-1归属为B酸中心吸附的NH4+中N-H键的不对称变形振动[26]。在NH3吸附过程中, 并未通入O2, 而红外光谱谱图上却出现了属于NH3氧化中间产物以及硝基类物种的特征峰, 推测这与催化剂本身的氧化性有关, 由于催化剂中含有一定的晶格氧, 可在无氧条件下, 进行氧化还原反应, 促进部分NH3反应[27]。当引入NO+O2后, 1624和1557 cm-1位置的峰很快消失, 1225 cm-1处的吸附峰也逐渐变小, 吸附态的NH3和中间产物-NH2发生了进一步的反应。同时, 出现了几处新的吸收峰, 1597 cm-1处为桥式硝酸盐物种;1518 cm-1为单齿硝酸盐、1415和1312 cm-1的单齿亚硝酸盐物种[24]。结果证明, 在150 ℃低温条件下, 催化剂Lewis酸中心吸附的NH3和-NH2物种与气相NO或弱吸附态的NO和NO2反应为主要的反应。
((a), (c))pre-adsorbed with NH3 and then exposed to NO+O2 for various times ((b), (d))pre-adsorbed with NO+O2 and then exposed to NH3 for various times
图 7(c)为M/0.5S/Al催化剂的FT-IR谱图。改性后催化剂预先吸附NH3的吸附峰位置与M/Al2O3催化剂基本相同。但在1463 cm-1处的吸附峰强度增高, 1225 cm-1处吸附峰减弱, 说明与M/0.5S/Al催化剂表面B酸中心吸附的NH4+较多, L酸吸附的NH3较少。当引入NO+O2后, 1557 cm-1处的NH2中间物种的峰很快消失, 1463 cm-1处的峰有所降低, 但1225 cm-1处吸附峰几乎没有变化。说明形成的中间物种和B酸位上少量的NH4+发生了反应, 但是由于催化剂的B强酸性较高, 导致L酸位上吸附的NH3不易脱附形成中间物种, 不易与气相NO反应[22]。同时, 在1391 cm-1处出现了一个吸附峰[26], 可能归属为硝酸盐, 并且随着NO+O2通入时间延长, 峰强度逐渐增加。说明在低温条件下, 改性催化剂上较多的B酸中心以及较弱的氧化能力, 导致NH3很难被活化参与反应, 因此,低温活性降低。
图 7(b)和(d)是先吸附NO+O2后, 再通NH3的FT-IR谱图。由图 7(b)可知, M/Al2O3催化剂在吸附NO+O2后, 在1611、1518、1415、1403和1312 cm-1处出现了不同的吸附峰。其中,1611 cm-1处振动峰归属于吸附态NO2[22, 28]或桥式硝酸盐[29], 1518和1312 cm-1处吸附峰归属于单齿硝基/亚硝基配体物种, 1403 cm-1可以归属为单齿亚硝酸盐的吸附峰。通入NH3后, 随着通入时间延长, 归属为单齿亚硝酸盐物种1312 cm-1处的吸附峰逐渐变小, 而其他峰变化不明显, 说明在此过程中NO2-物种可能与气相NH3或者弱吸附态的NH3发生反应, 双齿硝酸盐不利于反应的进行。而图 7(d)显示, M/0.5S/Al催化剂在1312 cm-1处的吸附峰很弱, 说明形成的亚硝基物质较少, 催化剂上硫的引入抑制了氮氧化物的吸附[21], NH3通入后, 吸附峰有微弱的减小, 说明反应非常少。
在150 ℃低温下, 催化剂上吸附的氨气或中间态氨可以与气相的NO或吸附态的NO发生反应;催化剂上吸附的亚硝酸盐, 可以与吸附态或中间态的NH3发生反应, 此过程所占比例较少。改性后催化剂, 由于氧化能力不足, B酸性增强, 仅发生少量吸附的氨气或中间态氨可以与气相的NO或吸附态的NO发生反应, 因此,脱硝能力较低。
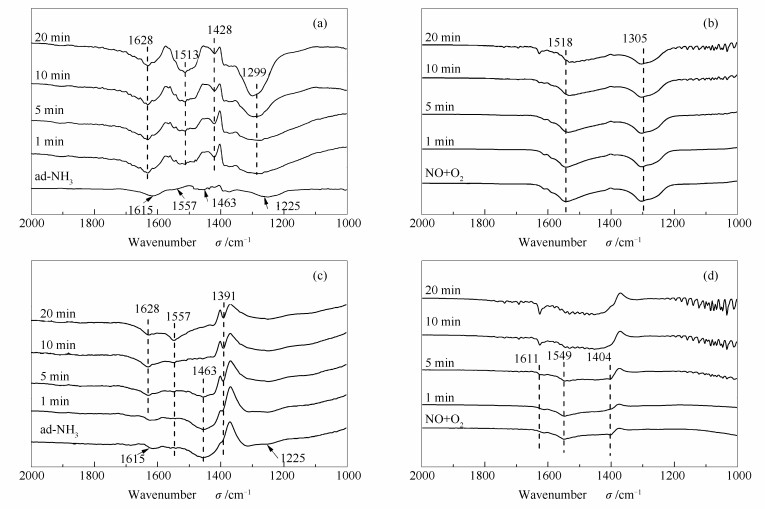
图 8为300 ℃下M/Al2O3和M/0.5S/Al催化剂的FT-IR谱图。图 8(a)和(c)为预先吸附NH3, 再通入NO+O2的过程。由图 8(a)可知, M/Al2O3催化剂吸附NH3后, 在1615、1557、1463和1225 cm-1处出现吸附峰。与150 ℃该催化剂上同样的吸附过程吸附峰位置相同, 但1557 cm-1处代表-NH2中间物种吸附峰和1225 cm-1归属为Lewis酸吸附NH3的对称变形振动峰较小。当通入NO+O2后, 1557 cm-1处代表-NH2中间物种吸附峰和1463 cm-1处代表B酸中心吸附的NH4+的峰消失;在1513、1428和1299 cm-1处出现新的吸收峰, 分别为硝酸盐和亚硝酸盐物种, 并且各位置的吸附峰强度大于150 ℃低温条件下的吸附峰。说明当催化剂氧化性较强时, 高温下NO容易被过度氧化, 被吸附形成硝酸盐或亚硝酸盐。
((a), (c))pre-adsorbed with NH3 and then exposed to NO+O2 for various times ((b), (d))pre-adsorbed with NO+O2 and then exposed to NH3 for various times
图 8(c)为M/0.5S/Al催化剂的FT-IR谱图。催化剂吸附NH3后, 在1615、1557、1463和1225 cm-1处出现不同吸附峰。当引入NO+O2后, 随着时间的延长, 1463 cm-1处的吸附峰明显减小至消失;在1391 cm-1处硝酸盐的吸附峰逐渐增加。说明在300 ℃条件下, 主要是催化剂表面B酸中心吸附的NH4+被活化参与反应进行[30, 31]。比较图 8(a)和(c)可以发现, 与未改性催化剂相比, 改性催化剂上, 硝酸盐类型发生变化, 并且硝酸盐的吸附峰面积明显较低。
图 8(b)和(d)是先通NO+O2, 再通NH3的过程。由图 8(b)可知, 通入NO+O2后, 催化剂在1518、1305 cm-1处出现了吸附峰, 分别归属为双齿硝酸盐和单齿亚硝酸盐。通入NH3后, 谱图几乎没有变化。同样, 图 8(d)中催化剂吸附NO+O2后, 形成的多种硝酸盐和亚硝酸盐的吸收峰, 随着NH3通入时间的延长, 吸附峰强度几乎没有太大变化, 说明此过程中NO被氧化形成硝酸盐后没有再进一步发生反应。因此, 在300 ℃条件下, 无论是改性前还是改性后的催化剂吸附态的硝酸盐或亚硝酸盐很难发生进一步反应。
在300 ℃条件下, 主要是气相的NO与B酸中心吸附的NH4+物种反应, 因此,M/0.5S/Al活性较高。
采用简单的硫酸改性载体的方法, 得到了具有不同B酸量的锰铈氧化铝催化剂, 研究了B酸性对催化剂SCR脱硝活性的影响。结果表明, 氧化铝载体改性, 降低了活性组分的分散及催化剂的氧化能力, 增强了载体的B酸量。改性液浓度越高, 催化剂酸性越强, 活性温度窗口越宽, 最高活性温度越高。当改性液浓度为0.2 mol/L, 催化剂在200-250 ℃的脱硝活性与未改性前基本相等, 但抗水抗硫性能明显改善。当烟气温度低于200 ℃时, 可选用未改性催化剂;当烟气温度在200-250 ℃时, 可选用0.2 mol/L浓度的改性液进行改性;当烟气温度高于300 ℃时, 可选用1 mol/L浓度的改性液进行改性。这就使锰铈类催化剂可应用于200-450 ℃的温度区间。
纪生晓, 张玮坚, 郑玉婴, 朱建风. 低温燃烧法制备Mn-CeOx催化剂及其NH3-SCR脱硝性能[J]. 燃料化学学报, 2019,47,(2): 224-235. doi: 10.3969/j.issn.0253-2409.2019.02.012JI Sheng-xiao, ZHANG Wei-jian, ZHENG Yu-ying, ZHU Jian-feng. Low-temperature combustion synthesis of the Mn-CeOx catalyst and its performance in the selective catalytic reduction of NOx by NH3[J]. J Fuel Chem Technol, 2019, 47(2): 224-235. doi: 10.3969/j.issn.0253-2409.2019.02.012
KANG M, PARK J H, CHOI J S, PARK E D, YIE J E. Low-temperature catalytic reduction of nitrogen oxides with ammonia over supported manganese oxide catalysts[J]. Korean J Chem Eng, 2007, 24(1): 191-195.
MOUSAVIA S M, PANAHI P N. Modeling and optimization of NH3-SCR performance of MnOx/γ-alumina nanocatalysts by response surface methodology[J]. J Taiwan Inst Chem E, 2016, 69: 68-77. doi: 10.1016/j.jtice.2016.09.033
郭静, 李彩亭, 路培, 崔华飞, 彭敦亮, 文青波. CeO2改性MnOx/Al2O3的低温SCR法脱硝性能及机制研究[J]. 环境科学, 2011(8): 2240-2246. GUO Jing, LI Cai-ting, LU Pei, CUI Hua-fei, PENG Dun-liang, WEN Qing-bo. Research on SCR denitrification of MnOx/Al2O3 modified by CeO2 and its mechanism at low temperature[J]. Environ sci, 2011, (8): 2240-2246.
JIN R B, LIU Y, WU Z B, WANG H Q, GU T T. Low-temperature selective catalytic reduction of NO with NH3 over Mn-Ce oxides supported on TiO2 and Al2O3:A comparative study[J]. Chemosphere, 2010, 78(9): 1160-1166. doi: 10.1016/j.chemosphere.2009.11.049
KANG M, PARK E D, KIM J M, YIE J E. Cu-Mn mixed oxides for low temperature NO reduction with NH3[J]. Catal Today, 2006, 111(3/4): 236-241.
QI G, YANG R T. Low-temperature selective catalytic reduction of NO with NH3 over iron and manganese oxides supported on titania[J]. App Catal B:Environ, 2003, 44(3): 217-225. doi: 10.1016/S0926-3373(03)00100-0
NOTOYA F, SU C, SASAOKA E, NOJIMA S. Effect of SO2 on the low-temperature selective catalytic reduction of nitric oxide with ammonia over TiO2, ZrO2, and Al2O3[J]. Ind Eng Chem Res, 2001, 40(17): 3732-3739. doi: 10.1021/ie000972f
XIE G, LIU Z, ZHU Z, LIU Q, GE J, HUANG Z. Simultaneous removal of SO2 and NOx from flue gas using a CuO/Al2O3 catalyst sorbent Ⅱ. Promotion of SCR activity by SO2 at high temperatures[J]. J Catal, 2004, 224(1): 42-49.
YAO X J, WANG Z, YU S H, YANG F, DONG L. Acid pretreatment effect on the physicochemical property and catalytic performance of CeO2 for NH3-SCR[J]. Appl Catal A:Gen, 2017, 542: 282-288. doi: 10.1016/j.apcata.2017.06.003
ZHANG L, ZOU W X, MA K L, CAO Y, XIONG Y, WU S G, TANG C J, GAO F, DONG L. Sulfated temperature effects on the catalytic activity of CeO2 in NH3-selective catalytic reduction conditions[J]. J Phys Chem C, 2015, 119(2): 1155-1163. doi: 10.1021/jp511282c
李晶. Mn基低温脱硝催化剂的研究[D].北京: 石油化工学院, 2015.LI Jing. Research on low-temperature Mn-based deNOx catalyst[D]. Beijing: Institute of Petrochemical Technology, 2015.
ZHAO W W, LI C T, LU P, WEN Q B, ZHAO Y P, ZHANG X, FAN C Z, TAO S S. Iron, lanthanumand manganese oxides loaded onγ-Al2O3 for selective catalytic reduction of NO with NH3 at low temperature[J]. Environ Technol, 2012, 34(1/4): 81-90.
黄增斌, 李翠清, 王振, 徐胜美, 冯凌波, 王虹, 宋永吉, 张伟. 不同分子筛负载锰铈催化剂的低温NH3-SCR脱硝性能[J]. 燃料化学学报, 2016,44,(11): 1388-1394. doi: 10.3969/j.issn.0253-2409.2016.11.016HUANG Zeng-bin, LI Cui-qing, WANG Zhen, XU Sheng-mei, FENG Ling-bo, WANG Hong, SONG Yong-ji, ZHANG Wei. Performance of Mn-Ce catalysts supported on different zeolites in the NH3-SCR of NOx[J]. J Fuel Chem Technol, 2016, 44(11): 1388-1394. doi: 10.3969/j.issn.0253-2409.2016.11.016
ETTIREDDY P R, ETTIREDDY N, MAMEDOV S, BOOLCHAND P, SMIRNIOTIS P G. Surface characterization studies of TiO2 supported manganese oxide catalysts for low temperature SCR of NO with NH3[J]. Appl Catal B:Environ, 2007, 76(1): 123-134.
安忠义, 禚玉群, 陈昌和. 煅烧温度对Mn/TiO2催化剂催化NO氧化活性的影响[J]. 燃料化学学报, 2014,42,(3): 370-377. AN Zhong-yi, ZHUO Yu-qun, CHEN Chang-he. Influence of calcination temperature on the catalytic activity of Mn/TiO2 for NO oxidation[J]. J Fuel Chem Technol, 2014, 42(3): 370-377.
张治安, 杨邦朝, 邓梅根, 胡永达, 汪斌华. 超级电容器纳米氧化锰电极材料的合成与表征[J]. 化学学报, 2004,62,(17): 1617-1620. doi: 10.3321/j.issn:0567-7351.2004.17.008ZHANG Zhi-an, YANG Bang-chao, DENG Mei-gen, HU Yong-da, WANG Bin-hua. Synthesis and characterization of nanostructured MnO2 for supercapacitor[J]. Acta Chim Sin(Chin Ed), 2004, 62(17): 1617-1620. doi: 10.3321/j.issn:0567-7351.2004.17.008
DU C H, QIN Y N, HE Y F, SHI X M, MA Z. Preparation and characterization of novel solid acid of sulfated anodized aluminium[J]. J Mol Catal, 2003, 17(3): 183-187.
HOSSEINPOUR N, MORTAZAVI Y, BAZYARI A, KHODADADI A A. Synergetic effects of Y-zeolite and amorphous silica-alumina as main FCC catalyst components on triisopropylbenzene cracking and coke formation[J]. Fuel Process Technol, 2009, 90(2): 171-179. doi: 10.1016/j.fuproc.2008.08.013
王斌, 张强, 韩东敏, 李春义, 杨朝合, 山红红. 催化剂基质Lewis及Bronsted酸性位强度对催化裂化小分子烯烃收率的影响[J]. 石油学报(石油加工), 2016,32,(4): 666-673. doi: 10.3969/j.issn.1001-8719.2016.04.002WANG Bin, ZHANG Qiang, HAN Dong-min, LI Chun-yi, YANG Chao-he, SHAN Hong-hong. Effects of acid strength of matrix in catalyst on the yield of small olefins during the catalytic cracking process[J]. Acta Pet Sin(Pet Process Sect), 2016, 32(4): 666-673. doi: 10.3969/j.issn.1001-8719.2016.04.002
MORALES A, AGUDELO M M R D, HERNÁNDEZ F. Adsorption mechanism of phosphorus on alumina[J]. Appl Catal, 1988, 41(41): 261-271.
ZHANG H, ZOU Y, PENG Y. Influence of sulfation on CeO2-ZrO2 catalysts for NO reduction with NH3[J]. Chin J Catal, 2017, 38(1): 160-167. doi: 10.1016/S1872-2067(16)62581-0
万义玲, 张传辉, 郭杨龙, 郭耘, 卢冠忠. CeO2-MnOx催化剂上氯乙烯有机废气的催化燃烧[J]. 催化学报, 2012,33,(3): 557-562. WAN Yi-ling, ZHANG Chuan-hui, GUO Yang-long, GUO Yun, LU Guan-zhong. Catalytic combustion of vinyl chloride emission over CeO2-MnOx catalyst[J]. Chin J Catal, 2012, 33(3): 557-562.
樊银明. Ce原位引入和负载于Mn/SAPO-34的低温NH3-SCR抗硫抗水性能与分子模拟研究[D].广州: 华南理工大学, 2017. http://cdmd.cnki.com.cn/Article/CDMD-10561-1018809426.htmFAN Yin-ming. Experimental and molecular simulation study on cerium presence in the framework and the surface of Mn/SAPO-34 resistance to SO2 and H2O in NH3-SCR at low temperature[D]. Guangzhou: South China University of Technology, 2017. http://cdmd.cnki.com.cn/Article/CDMD-10561-1018809426.htm
JIANG B Q, WU Z B, LIU Y, LEE S C, HO W K. DRIFT study of the SO2 effect on low-temperature SCR reaction over Fe-Mn/TiO2[J]. J Phys Chem C, 2010, 114(11): 4961-4965. doi: 10.1021/jp907783g
孙路石, 赵清森, 向军, 石金明, 王乐乐, 胡松, 苏胜. 原位漫反射红外光谱研究NO和NH3在CuO/Al2O3催化剂上的吸附行为[J]. 化工学报, 2009,60,(2): 444-450. doi: 10.3321/j.issn:0438-1157.2009.02.027SUN Lu-shi, ZHAO Qing-sen, XIANG Jun, SHI Jin-ming, WANG Le-le, HU Song, SU Sheng. Adsorption of NO and NH3 over CuO/γ-Al2O3 catalyst by DRIFTS[J]. CIESC J, 2009, 60(2): 444-450. doi: 10.3321/j.issn:0438-1157.2009.02.027
谢天.铝基锰系催化剂脱硫脱硝性能研究[D].武汉: 华中科技大学, 2011. http://www.wanfangdata.com.cn/details/detail.do?_type=degree&id=D187366XIE Tian. A thesis submitted in partial fulfillment of the requirements for the degree of master in engineering[D]. Wuhan: Huazhong University of Science and Technology, 2011. http://www.wanfangdata.com.cn/details/detail.do?_type=degree&id=D187366
QI G, YANG R T. Characterization and FT-IR studies of MnOx-CeO2 catalyst for low-temperature selective catalytic reduction of NO with NH3[J]. J Phys Chem B, 2004, 108(40): 15738-15747. doi: 10.1021/jp048431h
KIJLSTRA W S, BRANDS D S, SMIT H I, POELS E K, BLIEK A. Mechanism of the selective catalytic reduction of NO with NH3 over MnOx/Al2O3 Ⅱ. Reactivity of adsorbed NH3 and NO complexes[J]. J Catal, 1997, 171(1): 219-230.
TOPSOE N Y, TOPSOE H, DUMESIC J A. Vanadia/titania catalysts for selective catalytic reduction (SCR) of nitric-oxide by ammonia:Ⅰ. Combined temperature-programmed in-situ FT-IR and on-line Mass-Spectroscopy studies[J]. J Catal, 1995, 151(1): 226-240. doi: 10.1006/jcat.1995.1024
TOPSØE N Y. Mechanism of the selective catalytic reduction of nitric oxide by ammonia elucidated by In situ on-line fourier transform infrared spectroscopy[J]. Science, 1994, 265(5176): 1217-1219. doi: 10.1126/science.265.5176.1217

图 2 M/Al2O3和M/0.2S/Al2O3催化剂在250 ℃时的抗水抗硫性能
Figure 2 H2O and SO2 durability of M/Al2O3 and M/0.2S/Al2O3 for NH3-SCR of NO at 250 ℃

图 4 改性前后催化剂的NH3-TPD谱图
Figure 4 NH3-TPD profiles of the unmodified and modified catalysts

图 5 改性前后催化剂的Py-FTIR谱图
Figure 5 Py-FTIR spectra of the unmodified and modified catalysts

图 6 改性前后催化剂的H2-TPR谱图
Figure 6 H2-TPR profiles of the unmodified and modified catalysts

图 7 150 ℃反应条件下M/Al2O3((a), (b))和M/0.5S/Al((c), (d))的红外光谱谱图
Figure 7 In situ FT-IR spectra of M/Al2O3((a), (b)) and M/0.5S/Al((c), (d)) under different conditions at 150 ℃
((a), (c))pre-adsorbed with NH3 and then exposed to NO+O2 for various times ((b), (d))pre-adsorbed with NO+O2 and then exposed to NH3 for various times

图 8 300 ℃反应条件下M/Al2O3((a), (b))和M/0.5S/Al((c), (d))的红外光谱谱图
Figure 8 In situ FT-IR spectra of M/Al2O3((a), (b)) and M/0.5S/Al((c), (d)) under different conditions at 300 ℃
((a), (c))pre-adsorbed with NH3 and then exposed to NO+O2 for various times ((b), (d))pre-adsorbed with NO+O2 and then exposed to NH3 for various times
表 1 改性前后催化剂的物理化学性质
Table 1. Physico-chemical properties of the unmodified and modified catalysts
| Catalyst | Specific surface area A/(m2·g-1) | Average pore diameter d/nm | Pore volume v/(cm3·g-1) |
| Al2O3 0.2S/Al 0.5S/Al 1.0S/Al M/Al2O3 M/0.2S/Al M/0.5S/Al M/1.0S/Al |
177.3 168.1 164.3 165.2 143.2 132.1 139.1 136.3 |
3.61 3.58 3.59 3.61 3.62 4.29 3.63 3.93 |
0.24 0.25 0.25 0.25 0.21 0.21 0.22 0.21 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们