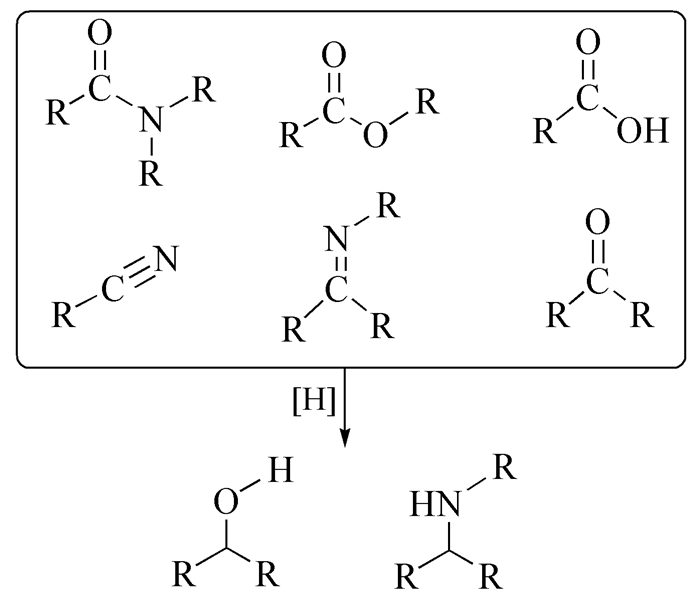
图式 1.
不饱和化合物的加氢反应
Scheme 1.
Hydrogenation reaction of unsaturated compounds

催化加氢/脱氢反应是有机催化中最基础的反应,从毫克级有机化学反应探索到吨级化学品的生产,均存在催化加氢和脱氢反应的广泛应用,同时也是研究得最深入的有机催化反应(常见的加氢/脱氢反应见图式 1和图式 2)。催化加氢反应原子经济性高,对环境的污染小,操作工艺简单,且催化加氢过程是放热反应,反应条件相对温和,较容易实现。催化脱氢反应是催化加氢反应的逆反应,其通过使用高效催化剂断裂有机物中的C—H键,同时还要维持着C—C键不断裂。常见的脱氢反应有醇类的脱氢、杂环的脱氢、烷烃的脱氢、氧化脱氢等[1~5]。
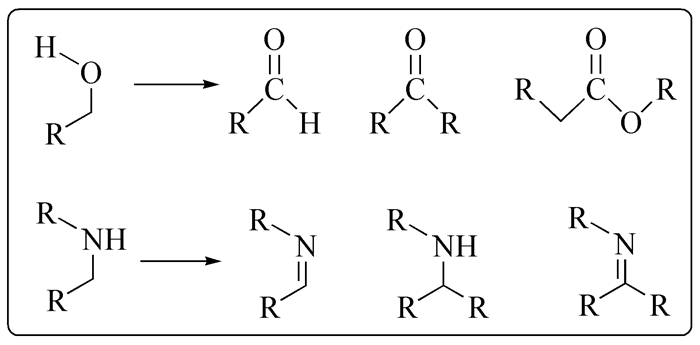
脱氢反应过程是一种吸热反应,且伴随着分子数的增加,因此通常需要高温高压的反应条件才能使脱氢顺利进行。过渡金属尤其是贵金属铑、钌、钯、铱等催化的加氢/脱氢反应取得了极大的发展,成为有机化学的重要研究方向。随着环保意识的增强及探求更加经济的催化方式,廉价金属铁、钴、锰等低毒且地球上储量丰富的元素在催化加氢/脱氢反应中的应用引起科学家们的极大关注。
本文主要从铁、钴、锰配合物催化的加氢/脱氢反应等方面进行介绍。
作为在地球上含量比较丰富的元素,在后过渡金属的研究中,铁配合物在均相还原催化中的应用相对比较成熟,很多综述介绍了铁催化剂在还原转化和有机合成中的反应性。
Morris等[6]报道了基于四齿亚氨基膦配体的铁配合物,并将其应用于酮的氢化反应研究中。该系列催化剂中比较具有代表性的是基于四齿PNNP配体的Fe配合物1,该配合物可在非常温和的条件(Fe催化剂量低于0.45(mol)%、50℃和25bar H2)下将苯乙酮高效还原为苯甲醇。而且,研究发现连接PNNP配体中N原子上基团的不同对Fe配合物催化剂的氢化反应活性有较大影响。
Milstein等[7]在铁配合物催化剂催化的酮的氢化反应方面取得了较大突破。他们开发的催化剂2在0.05(mol)%用量、室温和4.2×105 Pa H2压力下反应,即可实现与催化剂1相当的反应活性。铁催化剂2具有较广的底物适用范围,取代的苯乙酮、共轭二酮和含有C=C键的底物均能取得较好的反应结果。尽管大多数底物都以高收率转化,但是含有氨基和腈基官能团的底物反应活性较低。在不饱和酮的氢化中催化剂2只表现出中等的化学选择性,在反应完全时,底物中最多保留20%的C=C双键;另外,催化剂2在苯甲醛加氢反应中活性也较低,催化剂用量为0.125(mol)%时只得到36%的反应收率。
|
|
Kirchner等[8]报道了基于双官能团化配体的铁配合物3,并应用于氢化反应中。研究发现,在室温下、乙醇作为溶剂、5bar H2压力及0.5(mol)%的催化剂用量时,几乎可以定量还原酮和醛类底物。为了进一步提高该配合物的催化活性,他们进行了详尽的机理研究,而且实现了在酮基、酯基、环氧基、炔基和硝基芳族化合物存在条件下选择性还原醛类化合物。在机理指导下,优化了4-氟苯甲醛在(3~6)×106 Pa H2下的氢化反应,并得到最高达20000h-1的TOF值及80000的TON值,是目前报道的醛的选择性加氢活性最高的体系之一,可与贵金属催化体系相媲美。
|
|
Zirakzadeh等[9]报道的催化剂4(用量1(mol)%)在异丙醇中、2×106 Pa H2压力下可较好地实现酮的不对称氢化。Morris等[10]合成的催化剂5具有更高的催化活性,在催化剂量仅0.1(mol)%、1(mol)% KOtBu、50℃和106 Pa H2压力下,可实现取代苯乙酮的不对称氢化,ee值最高可达96%,而使用催化剂4的ee值最高仅为81%。
铁配合物在催化酯的氢化方面也表现出较好的反应活性。双氢-铁配合物6在40℃、(5~25)×105 Pa氢气条件下即可氢化活性酯类化合物[11]。NaOMe作为促进剂、0.5(mol)%~1(mol)%催化剂量下,一系列三氟乙酸酯均可以较好地完成转化。Gajewski等[12]利用催化剂7(1(mol)%)在7×106 Pa H2和90℃条件下实现了三氟乙酸酯的高效氢化。研究发现,三乙胺的存在对于催化反应至关重要,其可以中和反应过程中生成的三氟乙酸,减少它对催化剂的毒化作用。
|
|
非活性酯的氢化反应也取得了较大的研究进展,主要归功于钳型胺基配体骨架的发展及其铁配合物催化活性的提高。Beller等[13]和Guan等[14]分别利用催化剂8a(催化剂量1(mol)%~3(mol)%)在(1~5)×106 Pa氢气及100~130℃条件下实现了非活化酯的氢化反应。研究发现,8a的催化反应在无碱条件下即可实现,并且若在体系中加入含烷氧基的碱反而会抑制催化反应的活性。Beller等研究发现,位阻较小的配合物8b在催化酯的氢化反应时活性有所提高,表明催化剂的位阻效应一定程度上降低了催化剂的反应活性。
|
|
Beller等[15, 16]利用PNP铁配合物催化剂8a与8c实现了腈的加氢反应。在异丙醇中,30bar H2和70~100℃的条件下,无需碱的作用即可实现8a与8c催化的高效加氢反应。研究发现,若对钳形配体的NH基团进行甲基化,则会失去催化腈类氢化反应的活性。
Milstein等[17]利用双(2-二异丙基膦基苄基)胺配体与铁前体配位,形成具有铁中心的六元环金属配合物,生成的催化剂9仅有“PNP”配体的一个膦原子与金属铁中心配位。在催化剂9用量为1(mol)%~5(mol)%、NaHBEt3和双(三甲基硅烷基)氨基钾(KHMDS)为添加剂及140℃和6×106 Pa H2压力下,可将不同种类的脂肪腈和芳香腈类化合物转化为相应的胺。基于双膦配位的PNP铁配合物10在催化腈类化合物的氢化反应时得到的则是亚胺类产物。在3×106 Pa H2和90℃的条件下,催化剂10对亚胺产物的生成具有优异的化学选择性,成功保留C=N键,产物无过度还原[18]。
|
|
催化剂2不仅在酮的氢化反应中表现出优异的催化活性,Milstein等[19]研究发现,其在KHMDS的促进下对酰胺的加氢反应也表现出较高的反应活性。Langer等[20]也将Fe-胺基钳型配合物的应用拓展到了酰胺的氢化反应中。他们通过对配体取代基结构的筛选发现,取代基位阻效应较小的催化剂8b(用量2(mol)%~10(mol)%)在70~ 100℃及5×106 Pa H2无碱的条件下也可催化酰胺的氢化反应。具有类似结构的催化剂8c[21]在K3PO4的促进下也能实现酰胺的氢化反应,并且极大了提高了反应活性,不仅能实现个别底物的完全转化,而且催化剂量降低至0.33(mol)%,TON值为300~1000。Bernskoetter等[22]开发了一种五配位模式的铁-氢配合物11,其对于甲酰胺衍生物的氢化表现出超高的反应活性,在催化剂用量0.018(mol)%、3×106 Pa H2及100℃下,能实现一系列甲酰胺化合物的氢化,TON值最高可达4430。
与铁、锰金属催化体系相比,钴配合物催化的氢化反应研究相对较少。但是,目前所报道的钴配合物催化体系均具有较独特及优异的反应性能,进一步促进了廉价金属催化剂的发展进程。
Hanson等[23]报道了首例钴配合物催化剂对C=O及C=N键的氢化反应。二价钴配合物12在烯烃类、酮类及醛类化合物的氢化反应中活性均较高。在25~60℃、催化剂用量2(mol)%、1~4atm H2压力下,大部分底物均能达到90%以上转化率。
|
|
Kempe等[24]在钴催化的酮类化合物及醛类化合物的氢化方面取得了较大进展。催化剂13(用量0.25(mol)%~3(mol)%)在室温、2×106 Pa H2下可催化大部分醛和酮类化合物的氢化反应,均能达到较好的收率。而且,此催化体系具有较好的官能团耐受度,不同种类的芳基杂环化合物、卤代烃类化合物、与羰基共轭的C=C双键及远位置的C=C双键均能取得较好的反应结果。
|
|
Milstein等[25]报道了基于2, 6-二甲基吡啶钳型配体的钴配合物14,其配体为PNNH型,所含的二级胺官能团使得其催化酯类的氢化反应活性优于基于PNP或三级胺官能团的PNN配体催化剂的活性。催化剂用量2(mol)%~4(mol)%、8(mol)% NaBHEt3活化催化剂、KOtBu作为碱促进剂、5×106 Pa H2及130℃下,大部分产物能得到中等及以上收率。Jones等[26]则实现了更具有挑战性的酯类底物的氢化反应。利用催化剂15在5.5×106 Pa H2、120℃四氢呋喃溶剂中,可还原酯类化合物得到相应的醇类产物。
大部分钴催化的氢化反应是基于三齿或四齿的膦-给体配体,即钴金属前体与膦配体相互作用共同实现不同种类化合物的氢化反应。Elsevier等[27]利用原位反应的钴/三齿膦配体实现了羧酸类化合物及酯类化合物的氢化反应。在催化剂用量5(mol)%~10(mol)%、8×106 Pa H2、100℃下,可较好地将酯类化合物氢化,若底物中存在C=C双键,则双键也能被氢化;而且,更难被氢化的羧酸类化合物在无任何额外添加剂的条件下也可被氢化,例如,三氟乙酸在0.0125(mol)%催化剂量作用下即可完全转化,以50%的收率得到三氟乙醇。因此,该催化体系的催化性能与贵金属铱、铑催化体系相当。
|
|
催化剂14不仅能较好地催化酮类及醛类化合物的氢化反应,而且能应用于腈类化合物的氢化反应,但需要NaBHEt3活化催化剂、NaOtBu作为碱促进剂[28]。类似地,原位反应的催化体系17在腈类化合物氢化中也表现出较高的催化反应活性[29]。在100℃、5(mol)%催化剂量及3×106 Pa H2条件下,腈类化合物即可被定量还原为胺类化合物,芳基腈类与烷基腈类化合物均具有较好的反应活性。
从2016年开始,科学家们研究发现,基于钳型配体的锰配合物催化剂在极性键的氢化反应中表现出很高的反应活性,继而大量高活性的锰催化剂被开发并应用于氢化反应及其他有机合成反应中。
|
|
Beller等[30]首次利用基于胺基钳型配体的锰合物催化剂18实现了酮类、醛类化合物及芳基、烷基腈类化合物的氢化反应。与同类型的配合物相比,膦供体基团上带有异丙基取代基的催化剂18a具有更强的活性。在100~120℃、(3~5)×106 Pa H2和1(mol)%催化剂量下即可氢化酮类和醛类化合物;催化剂量为3(mol)%、NaOtBu作碱添加剂时可催化腈类化合物的氢化反应。他们还将锰配合物催化剂应用于酯的加氢催化反应中。然而,在120℃、8×106 P a H2压力下,催化剂18(用量2(mol)%)催化苯甲酸甲酯的加氢反应仅获得中等收率[31]。
Clarke等[32]开发的基于胺基钳型配体的锰催化剂19可用于酮类及酯类化合物的加氢反应,生成相应的醇类化合物。而且,反应可以在环境友好的醇类溶剂中进行,例如酮的氢化可以在乙醇中进行、酯类化合物则在异丙醇中反应,这对于钌等贵金属催化剂来说是比较大的挑战。
|
|
Kempe等[33]利用催化剂20实现了酮的加氢反应,这是目前催化活性比较高的体系之一,并且发现锰的价态对实现氢化反应至关重要。若将金属中心的Mn(Ⅰ)替换为Mn(Ⅱ),则催化反应不能进行;即使将Mn(Ⅱ)再次还原为Mn(Ⅰ),在无羰基的情况下,反应依旧不能进行。Beller等[34]利用催化剂21实现了酮的不对称氢化反应。在30~40℃、3×106 Pa H2、1(mol)%催化剂量下,不同种类的芳基酮及烷基酮均能转化为相应的醇类产物,并且具有较高的对映选择性。
|
|
与催化剂18结构类似的锰配合物21与22也能较好地催化酯的氢化反应,并且相对于催化剂18,反应条件更加温和。Pidko等[35]报道了无钳型配体骨架支撑的锰配合物23,发现[Mn(CO)2(PN)2]+在催化苯甲酸甲酯的氢化反应时活性低于三羰基的锰配合物Mn(PN)。在5×106 Pa H2、100℃、0.2(mol)%催化剂量下,即可催化酯的氢化反应进行。而且,距离酯基较远的C=C键在氢化反应中能得到保留;该氢化反应对于α, β-不饱和酯则没有化学选择性,双键与酯基同时被还原。
Milstein等[36, 37]报道了基于芳基骨架的锰配合物24,其在酯的氢化反应中表现出优异的催化反应活性。2×106 Pa H2、100℃、1(mol)%催化剂量下即可较好地实现不同酯类化合物的氢化反应。其中,碱对催化剂进行脱卤活化生成Mn-H活性中间体是反应能进行的关键。
铁配合物在催化酮类、酯类等化合物的氢化反应时,可以产生醇类产物,进一步研究发现,在其逆反应中,铁配合物催化剂也表现出优异的反应活性。Jones等[38]研究了铁配合物8a催化的N-杂环化合物的脱氢反应,并且发现与其结构十分类似的催化剂11不仅可以催化二级醇脱氢反应生成酮类化合物[39],还可以催化二级醇与一级醇类化合物的无受体脱氢偶联反应生成酯类产物,以及催化二醇类化合物分子内脱氢偶联生成内酯产物。
进一步研究发现,这类催化剂在强碱的作用下可以催化甘油脱氢生成乳酸盐类化合物[40],其中,8a与11在催化此类反应的催化剂中是目前活性最高的,0.004(mol)%~0.2(mol)%催化剂量下,最高可以得到1050的TON值。
|
|
胺类化合物在铁配合物的催化作用下,也可以发生脱氢偶联反应。Beller等[41]发现8a可以催化二醇或氨基醇的脱氢偶联反应实现内酯和内酰胺的合成[41]。Bernskoetter等[42]利用催化剂11催化伯醇(包括甲醇)与仲胺的脱氢酰胺化反应,生成相应的酰胺,TON值最高可达790。
|
|
基于芳基骨架的铁配合物也被应用于脱氢偶联反应中。Milstein等[43]通过催化剂10催化腈类化合物与胺类化合物的脱氢偶联反应,实现了醛亚胺的合成。在60℃、(1~2)×106 Pa H2和1(mol)%催化剂用量下可以选择性地合成各种芳基或烷基醛亚胺化合物。Kirchner等[44, 45]报道的基于三嗪及氨基吡啶骨架的Fe(Ⅱ)金属配合物3也可以催化醇与胺类化合物反应生成二级胺类化合物。
非钳型骨架的铁配合物7也是较好的脱氢偶联反应催化剂,Feringa等[46]将其应用于烷基或芳基胺类化合物与烷基或二醇类化合物的脱氢偶联反应,在120~130℃、1(mol)%催化剂量下即可实现烷基化,但是反应需要添加三甲胺N-氧化物作为活化剂。之后,他们将催化剂7的应用扩展到了伯胺、仲胺与苄醇衍生物的烷基化反应[47]以及由伯胺和不饱和二醇合成吡咯的反应中。
近年来,钴配合物催化剂也被应用于脱氢及脱氢偶联反应中。基于胺基骨架的配合物15作为潜在的脱氢催化剂[48],可以较好地实现醇与胺的脱氢偶联反应,生成亚胺产物、水及氢气。其中,醇脱氢是反应的起始步骤。Zhang等[49, 50]发现,若在此反应体系中加入分子筛,生成的产物变为二级胺类化合物。当使用2(mol)%的钴催化剂15时,包括芳族和脂族底物在内的多种醇和胺都可以以良好至优异的产率有效地转化为仲胺。具有芳基骨架的催化剂25也可以催化芳基及烷基醇类化合物与芳基胺类化合物的烷基化反应[51]。研究发现,不仅Co(Ⅱ)PCP可以催化反应进行,Co(Ⅲ)PCP也可以催化反应进行,只是反应活性相对较差。此外,Zhang等[52, 53]发现,催化剂15还可以催化胺与胺的选择性烷基化反应,生成多种仲胺,通过胺之间的交叉偶联或自身偶联,可将多种胺类底物转化为相应的仲胺,还可由相应的二胺前体合成环仲胺。
Kempe等[54~56]研究发现,催化剂26a、26b在相对温和的反应条件下,分别可以实现芳基胺与醇类的烷基化反应及二级醇与一级醇的烷基化反应。而且,在2.5(mol)%的催化剂量、100℃四氢呋喃中,催化剂26a还可以催化惰性酰胺的C-烷基化反应;催化剂26b在5(mol)%催化剂量下可以实现酯类化合物的烷基化反应。对于酰胺及酯类化合物的烷基化反应,体系中均需加入1.2~1.5倍化学计量的叔丁醇钾促进反应进行。
|
|
在实现锰催化的加氢反应以后,锰催化的脱氢及脱氢偶联反应也得到了快速的发展。催化剂18a、18b、22均可以催化醇类化合物对胺的烷基化反应[57]。异丙基取代的催化剂18a活性优于其他官能团取代的催化剂,尤其是在催化不同种类的芳基及烷基醇类试剂对芳基胺的烷基化反应中,几乎可以定量生成苄胺类化合物。此外,18a还可以应用于一级醇化合物对酮的α-烷基化[58]。
若锰催化的胺和醇的偶联反应未进行进一步的氢化反应,则反应生成醛亚胺类产物。Milstein等[59]报道了基于2, 6-二甲基吡啶的锰合物27,其在135℃、3(mol)%催化剂量下可催化醇与胺的脱氢偶联反应生成亚胺、氢气及水。虽然是在封闭体系中反应,但未发生进一步的加氢反应,产物停留在亚胺的阶段,表明此催化剂并不能催化C=N键的加氢反应。
Kirchner等[60, 61]利用配合物28催化甲醇与胺类化合物的脱氢偶联反应,实现了胺类化合物的甲基化。该反应条件较温和,不同种类的胺均能较好地反应,进一步拓展了脱氢偶联反应的应用范围。
|
|
对于廉价金属催化的作用机理目前还在进一步的探索中。但对于反应路径,普遍认为其经历了配体与金属中心的协同活化作用。例如,对于基于二甲基吡啶的金属配合物,吡啶基的亚甲基侧臂Ar-CH2提供了活性位点,其可以被强碱去质子化,从而导致配体去芳构化并形成配位不饱和配合物,进而金属中心可以与氢气作用,生成[M-H]物种。类似地,基于胺基的钳型金属配合物的NH结构去质子化形成五配位的模式,其中NH结构转化为与金属键合的酰胺,再与氢气作用。正是这种可逆的与氢气的作用过程,实现了廉价金属催化的脱氢/加氢反应路径。
|
|
综上所述,廉价金属作为传统贵金属的替代品,不仅具有低毒、价格低廉的优点,而且表现出不逊于贵金属催化剂的催化活性,进而成为近年来的研究热点。本文主要介绍了目前报道较多的廉价铁、钴、锰等配合物催化的加氢、脱氢及脱氢偶联反应,此类催化剂的应用为合成不同类型的酮类、酯类、胺类化合物及各种类型的杂环化合物提供了更加低成本、简便、快捷的方法。
Alig L, Fritz M, Schneider S. Chem. Rev., 2019, 119(4): 2681~2751. doi: 10.1021/acs.chemrev.8b00555
Gunanathan C, Milstein D. Science, 2013, 341(6143): 1229712. doi: 10.1126/science.1229712
Kumar A, Bhatti T M, Goldman A S. Chem. Rev., 2017, 117(19): 12357~12384. doi: 10.1021/acs.chemrev.7b00247
Crabtree R H. Chem. Rev., 2017, 117(13): 9228~9246. doi: 10.1021/acs.chemrev.6b00556
Filonenko G A, van Putten R, Hensen E J M, et al. Chem. Soc. Rev., 2018, 47: 1459~1483. doi: 10.1039/C7CS00334J
Sui-Seng C, Haque F N, Hadzovic A, et al. Inorg. Chem., 2009, 48(2): 735~743. doi: 10.1021/ic801518h
Langer R, Leitus G, Ben-David Y, et al. Angew. Chem. Int. Ed., 2011, 50(9): 2120~2124. doi: 10.1002/anie.201007406
Langer R, Diskin-Posner Y, Leitus G, et al. Angew. Chem. Int. Ed., 2011, 50(42): 9948~9952 doi: 10.1002/anie.201104542
Zirakzadeh A, Kirchner K, Roller A, et al. Organometallics, 2016, 35(21): 3781~3787. doi: 10.1021/acs.organomet.6b00711
Smith S A M, Lagaditis P O, Lüpke A, et al. Chem. Eur. J., 2017, 23(30): 7212~7216. doi: 10.1002/chem.201701254
Zell T, Ben-David Y, Milstein D. Angew. Chem. Int. Ed., 2014, 53(18): 4685~4689. doi: 10.1002/anie.201311221
Gajewski P, Gonzalez-de-Castro A, Renom-Carrasco M, et al. ChemCatChem, 2016, 8(22): 3431~3435. doi: 10.1002/cctc.201600972
Werkmeister S, Junge K, Wendt B, et al. Angew. Chem. Int. Ed., 2014, 53(33): 8722~8726. doi: 10.1002/anie.201402542
Chakraborty S, Dai H, Bhattacharya P, et al. J. Am. Chem. Soc., 2014, 136(22): 7869~7872. doi: 10.1021/ja504034q
Lange S, Elangovan S, Cordes C, et al. Catal. Sci. Technol., 2016, 6: 4768~4772. doi: 10.1039/C6CY00834H
Bornschein C, Werkmeister S, Wendt B, et al. Nat. Commun., 2014, 5: 4111. doi: 10.1038/ncomms5111
Chakraborty S, Leitus G, Milstein D. Chem. Commun., 2016, 52: 1812~1815. doi: 10.1039/C5CC08204H
Chakraborty S, Milstein D. ACS Catal., 2017, 7(6): 3968~3972. doi: 10.1021/acscatal.7b00906
Garg J A, Chakraborty S, Ben-David Y, et al. Chem. Commun., 2016, 52: 5285~5288. doi: 10.1039/C6CC01505K
Schneck F, Assmann M, Balmer M, et al. Organometallics, 2016, 35(11): 1931~1943. doi: 10.1021/acs.organomet.6b00251
Rezayee N M, Samblanet D C, Sanford M S. ACS Catal., 2016, 6(10): 6377~6383. doi: 10.1021/acscatal.6b01454
Jayarathne U, Zhang Y, Hazari N, et al. Organometallics, 2017, 36(2): 409~416. doi: 10.1021/acs.organomet.6b00816
Zhang G, Scott B L, Hanson S K. Angew. Chem. Int. Ed., 2012, 51(48): 12102~12106. doi: 10.1002/anie.201206051
Rösler S, Obenauf J, Kempe R. J. Am. Chem. Soc., 2015, 137(25): 7998~8001. doi: 10.1021/jacs.5b04349
Srimani D, Mukherjee A, Goldberg A F G, et al. Angew. Chem. Int. Ed., 2015, 54(42): 12357~12360. doi: 10.1002/anie.201502418
Yuwen J, Chakraborty S, Brennessel W W, et al. ACS Catal., 2017, 7(5): 3735~3740. doi: 10.1021/acscatal.7b00623
Korstanje T J, van der Vlugt J I, Elsevier C J, et al. Science, 2015, 350(6258): 298~302. doi: 10.1126/science.aaa8938
Mukherjee A, Srimani D, Chakraborty S, et al. J. Am. Chem. Soc., 2015, 137(28): 8888~8891. doi: 10.1021/jacs.5b04879
Adam R, Bheeter C B, Cabrero-Antonino J R, et al. ChemSusChem, 2017, 10(5): 842~846. doi: 10.1002/cssc.201601843
Elangovan S, Topf C, Fischer S, et al. J. Am. Chem. Soc., 2016, 138(28): 8809~8814. doi: 10.1021/jacs.6b03709
Elangovan S, Garbe M, Jiao H, et al. Angew. Chem. Int. Ed., 2016, 55(49): 15364~15368. doi: 10.1002/anie.201607233
Widegren M B, Harkness G J, Slawin A M Z, et al. Angew. Chem. Int. Ed. 2017, 56(21): 5825~5828. doi: 10.1002/anie.201702406
Kallmeier F, Irrgang T, Dietel T, et al. Angew. Chem. Int. Ed., 2016, 55(39): 11806~11809. doi: 10.1002/anie.201606218
Garbe M, Junge K, Walker S, et al. Angew. Chem. Int. Ed., 2017, 56(37): 11237~11241. doi: 10.1002/anie.201705471
van Putten R, Uslamin E A, Garbe M, et al. Angew. Chem. Int. Ed., 2017, 56(26): 7531~7534. doi: 10.1002/anie.201701365
Espinosa-Jalapa N A, Nerush A, Shimon L J W, et al. Chem. Eur. J., 2017, 23(25): 5934~5938. doi: 10.1002/chem.201604991
Fu S, Shao Z, Wang Y, et al. J. Am. Chem. Soc., 2017, 139(34): 11941~11948. doi: 10.1021/jacs.7b05939
Chakraborty S, Brennessel W W, Jones W D. J. Am. Chem. Soc., 2014, 136(24): 8564~8567. doi: 10.1021/ja504523b
Chakraborty S, Lagaditis P O, Förster M. ACS Catal., 2014, 4(11): 3994~4003. doi: 10.1021/cs5009656
Sharninghausen L S, Mercado B Q, Crabtree R H, et al. Chem. Commun., 2015, 51: 16201~16204. doi: 10.1039/C5CC06857F
Peña-López M, Neumann H, Beller M. ChemCatChem, 2015, 7(5): 865~871. doi: 10.1002/cctc.201402967
Lane E M, Uttley K B, Hazari N, et al. Organometallics, 2017, 36(10): 2020~2025. doi: 10.1021/acs.organomet.7b00258
Chakraborty S, Leitus G, Milstein D. Angew. Chem. Int. Ed., 2017, 56(8): 2074~2078. doi: 10.1002/anie.201608537
Mastalir M, Stöger B, Pittenauer E, et al. Adv. Synth. Catal., 2016, 358(23): 3824~3831. doi: 10.1002/adsc.201600689
Mastalir M, Glatz M, Gorgas N, et al. Chem. Eur. J., 2016, 22(35): 12316~12320. doi: 10.1002/chem.201603148
Yan T, Feringa B L, Barta K. Nat. Commun., 2014, 5: 5602. doi: 10.1038/ncomms6602
Yan T, Feringa B L, Barta K. ACS Catal., 2016, 6(1): 381~388. doi: 10.1021/acscatal.5b02160
Zhang G, Vasudevan K V, Scott B L, et al. J. Am. Chem. Soc., 2013, 135(23): 8668~8681. doi: 10.1021/ja402679a
Zhang G, Hanson S K. Org. Lett., 2013, 15(3): 650~653. doi: 10.1021/ol303479f
Zhang G, Yin Z, Zheng S. Org. Lett., 2016, 18(2): 300~303. doi: 10.1021/acs.orglett.5b03461
Mastalir M, Tomsu G, Pittenauer E, et al. Org. Lett. 2016, 18(14): 3462~3465. doi: 10.1021/acs.orglett.6b01647
Zhang G, Wu J, Zeng H, et al. Org. Lett. 2017, 19(5): 1080~1083. doi: 10.1021/acs.orglett.7b00106
Yin Z, Zeng H, Wu J, et al. ACS Catal., 2016, 6(10): 6546~6550. doi: 10.1021/acscatal.6b02218
Rösler S, Ertl M, Irrgang T, et al. Angew. Chem. Int. Ed., 2015, 54(50): 15046~15050. doi: 10.1002/anie.201507955
Freitag F, Irrgang T, Kempe R. Chem. Eur. J., 2017, 23(50): 12110~12113. doi: 10.1002/chem.201701211
Deibl N, Kempe R. J. Am. Chem. Soc., 2016, 138(34): 10786~10789. doi: 10.1021/jacs.6b06448
Elangovan S, Neumann J, Sortais J B, et al. Nat. Commun., 2016, 7: 12641. doi: 10.1038/ncomms12641
Peña-López M, Piehl P, Elangovan S, et al. Angew. Chem. Int. Ed., 2016, 55(48): 14967~14971. doi: 10.1002/anie.201607072
Mukherjee A, Nerush A, Leitus G, et al. J. Am. Chem. Soc., 2016, 138(13): 4298~4301. doi: 10.1021/jacs.5b13519
Mastalir M, Pittenauer E, Allmaier G, et al. J. Am. Chem. Soc., 2017, 139(26): 8812~8815. doi: 10.1021/jacs.7b05253
Schlagbauer M, Kallmeier F, Irrgang T, et al. Angew. Chem. Int. Ed., 2020, 59(4): 1485~1490. doi: 10.1002/anie.201912055

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
