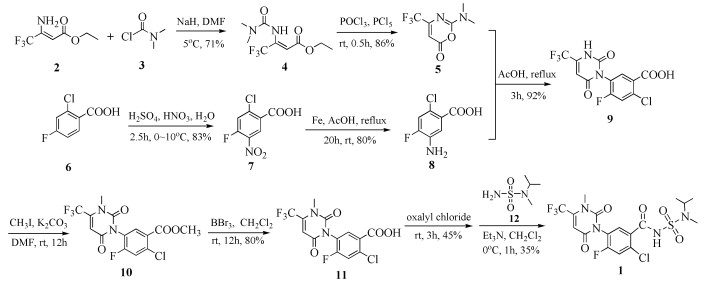
图式 1.
苯嘧磺草胺(1)原研合成路线
Scheme 1.
Preliminary synthetic route of saflufenacil (1)

苯嘧磺草胺(Saflufenacil)是巴斯夫公司研制并开发的脲嘧啶类除草剂,化学名称为N″-[2-氯-4-氟-5-(3-甲基-2, 6-二酮-4-(三氟甲基)-3, 6-二氢-1(2H)嘧啶基)苯甲酰基]-N-异丙基-N-甲磺酰胺[1]。其作用机理是通过抑制原卟啉原氧化酶(PPO)来实现除草功效[2~4]。通过妨碍叶绿素生物合成作用,可有效防除玉米、高粱、大豆、小粒谷物、棉花、果树和坚果中70多种杂草,且对作物非常安全,具有低挥发性、“有利的”毒理学和生态学特性。由于其在除草方面的卓越表现,苯嘧磺草胺被巴斯夫公司誉为“二十多年来开发最成功的新除草剂”[5, 6]。
如图式 1所示,苯嘧磺草胺的原研合成路线[7]以3-氨基-4, 4, 4-三氟巴豆酸乙酯(2)和N, N-二甲氨基甲酰氯(3)为原料,经过NaH/DMF条件酰胺化、POCl3/PCl5分子内成环得到脲嘧啶环化合物5,两步反应总收率61%;以2-氯-4-氟苯甲酸(6)为原料经过硝化反应得到2-氯-4-氟-5-硝基苯甲酸(7),再经还原反应得到5-氨基-2-氯-4-氟苯甲酸(8),两步反应总收率66%;脲嘧啶化合物5与苯甲酸化合物8反应,将脲嘧啶环与苯系化合物偶联得到2-氯-5-(2, 6-二氧-4-(三氟甲基)-3, 6-二氢嘧啶-1(2H)-基)-4-氟苯甲酸(9),再经过碘甲烷发生双甲基化得到苯甲酸甲酯化合物10,而后用三溴化硼水解为苯甲酸化合物11,两步反应总收率70%;最后将化合物11与草酰氯反应制备成酰氯、再与N-甲基-N-异丙基氨基磺酰胺(12)缩合得到目标产物苯嘧磺草胺,后两步总收率约16%。
该路线的思路为先期构建脲嘧啶环,经甲基化之后再与侧链N-甲基-N-异丙基胺基磺酰胺(12)缩合得到最终产物苯嘧磺草胺。先期构建脲嘧啶环后,还要经过酰化反应制备酰氯,再经过缩合反应制备酰胺,反应条件比较剧烈。该路线合成方式很繁琐且合成步骤较多、总收率低,不适合大规模工业化生产。
文献报道的N-甲基-N-异丙基胺基磺酰胺(12)合成目前有如下几条路线,路线一[8]:采用N-异丙基甲胺和磺酰氯进行缩合反应,再使用氨气对其进行氨化得到化合物12。该路线缩合反应过程选择性较差,会产生二次缩合产物,反应收率较低;反应使用氨气,需在高压反应釜中进行,条件比较苛刻;路线二[7]:使用2, 4, 5-三氯苯酚对氨基磺酰氯进行保护,生成2, 4, 5-三氯苯磺酰胺,再和N-异丙基甲胺发生酯的氨解反应得到化合物12。由于2, 4, 5-三氯苯酚市售价格比较高,增加生产成本,其作为保护基难以回收,原子经济性差,最终产物需经柱层析纯化,不适合放大生产。
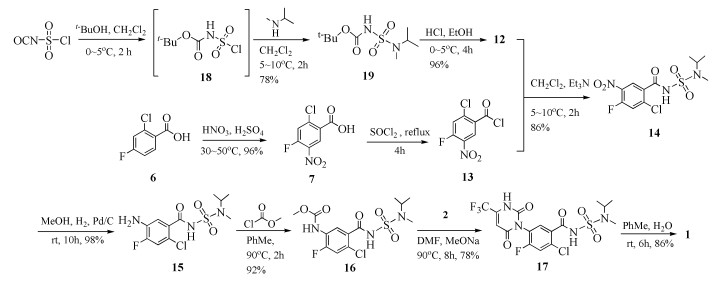
本文在上述路线的基础上进行了改进(反应路线见图式 2),以氯磺酰异氰酸酯为原料,首先使用叔丁醇进行氨基保护,再和N-异丙基甲胺缩合得到中间产物叔丁基(N-异丙基-N-甲基磺酰基)氨基甲酸酯(19),两步总收率78%;化合物19经氯化氢乙醇溶液脱除叔丁氧羰基得到化合物12,收率96%。在此基础上,以2-氯-4-氟苯甲酸(6)为原料,首先在混酸条件下进行硝化反应,得到中间体2-氯-4-氟-5-硝基苯甲酸(7),收率96%;而后使用二氯亚砜进行酰氯化得到苯甲酰氯化合物13,后者与化合物12进行酰胺化反应,得到中间体2-氯-4-氟-N-(N-异丙基-N-甲基)磺酰基-5-硝基苯甲酰胺(14),收率86%;再经催化氢化还原其硝基得到化合物15,收率98%;苯胺化合物15与氯甲酸甲酯缩合反应得到化合物16,收率92%;后者与3-氨基-4, 4, 4-三氟巴豆酸乙酯(2)经酯的氨解、分子内成环得到了脲嘧啶环化合物17,收率78%。最后使用硫酸二甲酯在弱酸性条件下进行甲基化得到目标产物苯嘧磺草胺1,收率86%。
本文在制备得到化合物12后,先与苯甲酰氯化合物13反应制备得到化合物14,反应条件温和,收率达到86%。其后续的催化氢化、氨基酰化、分子内成环、甲基化各步骤均能以较高收率实现。相比于原研路线采用的策略,最后由化合物11经酰氯化后再与化合物12反应制备得到目标产物(收率约16%)相比,本文的路线收率具有明显优势。
实验所使用化学药品和溶剂均为市售分析纯级,未经无水处理和进一步纯化。
Bruker UltraShield 400型核磁共振谱仪;岛津LC-16高效液相色谱仪。
称取10.0g(0.07mol)氯磺酰异氰酸酯置于250mL三口瓶中,加入60mL二氯甲烷,冰水浴搅拌,控制反应体系温度在5~10 ℃。将5.5g(0.074mol)叔丁醇用10mL二氯甲烷稀释,然后将其缓慢滴加入三口瓶中,滴加完毕,保温搅拌反应2h。向上述反应体系中加入10.6g(0.104mol)三乙胺,称取5.4g(0.074mol) N-异丙基甲胺并用10mL二氯甲烷稀释,然后将其缓慢滴加入上述三口瓶中,保持内温5~10 ℃,继续搅拌反应2h,反应完毕,反应液过滤除去无机盐,水洗涤滤液80mL×3次,收集有机相,使用无水硫酸钠干燥,过滤,旋蒸得到13.9g黄色油状产物叔丁基(N-异丙基-N-甲基磺酰基)氨基甲酸酯(19),收率78.7%。1H NMR (400MHz,DMSO-d6)δ:10.90(s,1H),3.95(m,1H),2.74(s,3H),1.42(s,9H),1.08(d,J=8.0Hz,6H)。
向上述所得黄色油状物中加入20mL乙醇,冰浴搅拌,滴加60mL 2mol/L氯化氢乙醇溶液,冰浴条件反应4h。反应结束,旋蒸除去大部分溶剂,向残余液中加入15mL甲基叔丁基醚,冰浴搅拌析晶,抽滤、烘干得白色固体8.0g,收率96%。熔点52.7~54.2 ℃(文献值51~53 ℃[9])。1H NMR (400MHz,DMSO-d6)δ:6.60(br s,1H),3.95(m,1H),2.55(s,3H),1.07(d,J=8.0Hz,6H)。
量取40mL浓硫酸(98%)加入三口瓶中,将其置于冰水浴中搅拌,称取10g(57.27mmol)2-氯-4-氟苯甲酸搅拌下缓慢加入上述硫酸中。称取8.6g(88.72mmol)65% HNO3缓慢滴加到三口瓶中,滴加过程逐渐有大量白色固体析出。滴加完毕,将反应液加热到50℃,搅拌反应2h。TLC监测至反应结束,将反应液倒入200mL冰水中,搅拌,析出固体,抽滤,并用水洗滤饼3次,45℃烘干得到白色固体12.1g,收率96%。熔点148.7~149.5 ℃(文献值150~153 ℃[10])。1H NMR(400MHz,CDCl3)δ:8.86(d,J=8.0Hz,1H),7.53(d,J=8.0Hz,1H)。MS(API) m/z:219.9 [M+H]+。
HPLC归一化法:色谱柱:Agilent Eclipse XDB-C18(250mm×4.6mm×5μm);检测波长:254nm;流速:0.8mL/min;柱温:35℃;进样量:1μL;溶剂:MeOH;浓度:0.5mg/mL;运行时间:20min;流动相:甲醇/水(80/20);保留时间(tR):3.017min,纯度:99.56%。
称取10.0g(0.045mol)化合物7加入茄型瓶中,加入50mL二氯亚砜,将其加热至80℃回流4h,反应结束,旋蒸除去溶剂,然后加入20mL甲苯继续旋蒸,以除去残留的二氯亚砜。冷却至室温得到白色固体10.5g,收率97%。熔点138.7~143.4℃。直接用于下一步反应。
称取6.7g(0.04mol)合成的化合物12置于三口瓶中,加入50mL二氯甲烷,冰水浴控制反应体系温度在5~10℃,称取4.5g(0.04mol)三乙胺加入到反应体系中,搅拌均匀。将10.0g (0.04mol)合成的化合物13用100mL二氯甲烷溶解,然后缓慢将其滴加进三口瓶中,滴加过程注意控制反应体系温度在5~10℃,滴加完毕,保温搅拌2h,TLC显示反应结束,抽滤除去三乙胺盐酸盐,收集滤液,并用水洗涤滤液(100mL×3次),分液,有机层经无水硫酸钠干燥,旋蒸得淡黄色固体12.8g,收率86%。熔点118.4~120.8 ℃。1H NMR (400MHz,CDCl3)δ:8.84 (br s,1H),8.52 (d,J=8.0Hz,1H),7.46(d,J=12.0Hz,1H),4.26~4.32 (m,1H),2.96(s,3H),1.23(d,J=8.0Hz,6H)。13C NMR (101MHz,DMSO-d6)δ:163.4,156.6,153.9,138.0(d,J=11.0Hz),135.8(d,J=7.8Hz),132.5,127.2,121.0(d,J=24.4Hz),49.4,28.6,20.1;MS (API) m/z:354.0 [M+H]+。
HPLC条件:柱型号:Acclaim C18 (150mm×2.1mm×5μm);检测波长:254nm;流速:0.8mL/min;柱温:35℃;进样量:1μL;溶剂:MeOH;时间:20min;流动相:甲醇/水=80/20,tR:3.309min,纯度:99.88%。
称取10.0g(0.028mol)化合物14置于茄型瓶中,加入80mL甲醇搅拌溶解,称取0.5g湿Pd/C(5%)加入茄型瓶中,通入氢气,并置换空气3次,室温反应10h,反应完毕,反应液使用硅藻土过滤除去钯碳,并用甲醇冲洗滤饼3次,旋蒸除去甲醇得到淡黄色固体9.0g,收率98.4%。熔点158.4~160.5℃。1H NMR (400MHz,DMSO-d6)δ:11.91(br s,1H),7.26 (d,J=8.0Hz,1H),6.85(d,J=8.0Hz,1H),5.60(s,2H),4.10~4.06(m,1H),2.81(s,3H),1.12(d,J=8.0Hz,6H)。13C NMR(101MHz,DMSO-d6)δ:165.3,152.3,149.9,136.3(d,J=12.9Hz),131.0(d,J=2.3Hz),117.0(d,J=2.3Hz),116.1(d,J=6.0Hz),115.5(d,J=9.5Hz),49.4,28.7,20.0。MS (API) m/z:324.0 [M+H]+。
HPLC条件:柱型号:Acclaim C18 (150mm×2.1mm×5μm);检测波长:254nm;流速:0.8mL/min;柱温:35℃;进样量:1μL;溶剂:MeOH;时间:20min;流动性:甲醇/水=80/20,tR:3.886min,纯度:99.90%。
称取6.5g(0.02mol)化合物15置于三口瓶中,加入100mL甲苯,加热使其溶解,升温至90℃,滴加2.8g(0.03mol)氯甲酸甲酯,保温反应2h,TLC显示反应完毕,自然冷却至室温,旋蒸除去甲苯、多余的氯甲酸甲酯以及生成的氯化氢气体,得到棕色固体7.0g,收率92%。熔点125.7~128.8℃。1H NMR (400MHz,DMSO-d6)δ:11.90(s,1H),9.58(s,1H),8.24(d,J=8.0Hz,1H),7.37(m,1H),4.33~4.27(m,1H),3.70(s,3H),2.83(s,3H),1.08(d,J=4.0Hz,6H)。13C NMR (101MHz,DMSO-d6)δ:165.0,154.8,129.0,127.0(d,J=11.3Hz),125.9(d,J=8.2Hz),124.9,116.4,116.2,52.6,49.3,28.8,19.9。MS(API) m/z:382.0 [M+H]+,348.1 [M-Cl]+。
HPLC条件:柱型号:Acclaim C18 (150mm×2.1mm×5μm);检测波长:220nm;流速:0.8mL/min;柱温:35℃;进样量:1μL;溶剂:MeOH;时间:15min;流动性:甲醇/水=80/20,tR:3.567min,纯度:98.86%。
称取6.5g(0.02mol)化合物16置于250mL三口瓶中,加入45mL DMF搅拌溶解。称取3.4g(0.02mol)化合物2加入三口瓶中,体系升温至90℃。将7.3g(0.04mol)30%甲醇钠甲醇溶液缓慢滴加到反应体系中,保温反应8h。反应完毕,将反应液自然冷却至室温,然后将其倒入200mL冰水中,并用浓盐酸调节反应液pH<2,逐渐有固体析出,室温搅拌30min,抽滤,并用水洗涤滤饼3次,得到化合物17 (7.0g),收率为78%。熔点228.2~229.3℃。1H NMR (400MHz,DMSO-d6)δ:12.00(s,1H),8.08(m,2H),7.56(t,J=8.0Hz,1H),6.44(s,1H),4.15(m,1H),2.84(s,3H),1.10(d,J=8.0Hz,6H)。13C NMR(101MHz,DMSO-d6)δ:164.4,161.7,159.3,150.5,132.3,131.7(d,J=9.4Hz),129.6(d,J=3.8Hz),123.0(d,J=3.8Hz),121.1,116.9,116.7,100.7(d,J=3.9Hz),49.4,28.8,19.9。MS (API):m/z=453.0 [M-Cl]+。
HPLC条件:柱型号:Acclaim C18 (150mm×2.1mm×5μm);检测波长:220nm;流速:0.8mL/min;柱温:35℃;进样量:1μL;溶剂:MeOH;时间:20min;流动性:甲醇/水=80/20,tR:2.981min,纯度:99.02%。
称取6.5g(0.01mol)化合物17置于三口瓶中,加入25mL甲苯、10mL四氢呋喃和15mL水,再加入0.2g(3.3(mol)%)四丁基溴化铵搅拌均匀,称取2.1g(0.017mol)硫酸二甲酯缓慢滴加进三口瓶中。反应过程不断补加10% NaOH溶液控制反应pH=4~5,搅拌反应6h。反应结束,分液,收集甲苯层20mL水洗3次,分液,无水硫酸钠干燥,旋蒸除去甲苯得到产品5.75g,产率86.1%。熔点189.2~191.4℃。1H NMR (400MHz,DMSO-d6)δ:12.03 (s,1H),8.02(d,J=7.2Hz,1H),7.54(t,J=7.2Hz,1H),6.59(s,1H),4.11(m,1H),3.43(s,3H),2.80(s,3H),1.08(d,J=6.8Hz,6H)。13C NMR(101MHz,DMSO-d6) δ:163.0,161.9,160.2,159.3,150.8,142.0(d,J=34.3Hz),131.3(d,J=9.0Hz),130.5,128.8(d,J=4.0Hz),122.4(d,J=14.0Hz),117.2(d,J=21.0Hz),103.0,49.9,32.8,28.6,19.9。MS (API):m/z=467.1 [M-Cl]+。HRMS m/z:C17H17ClF4N4O5S:理论值499.0466;实测值499.0470[M-H]。
HPLC条件:柱型号:Acclaim C18 (150mm×2.1mm×5μm);检测波长:254nm;流速:0.8mL/min;柱温:30℃;进样量:1μL;溶剂:MeOH;浓度:0.2mg/mL;时间:20min;流动性:甲醇/水=80/20,tR:3.598min,纯度:98.66%。
考察了投料比、反应温度、缚酸剂、溶剂对反应收率的影响,最终确定优化反应条件为:采用氯磺酰异氰酸酯为起始原料,氯磺酰异氰酸酯、叔丁醇与N-异丙基甲基胺摩尔比为1:1.06:1.06,二氯甲烷作溶剂,反应温度5℃,1.5倍量三乙胺作缚酸剂。使用HCl/乙醇溶液脱Boc得到化合物12,收率78%。
苯嘧磺草胺化学结构可以拆分为脲嘧啶环,含氟、氯的苯环以及N-甲基-N-异丙基氨基磺酰胺。整个分子结构设计的关键点在于脲嘧啶环的构建以及侧链N-甲基-N-异丙基氨基磺酰胺的合成,同时通过查阅相关文献[7, 10]发现,侧链12和脲嘧啶接入的先后顺序对反应过程也有影响。本文采用先期连接侧链结构,之后再进行脲嘧啶环构建的策略,该路线较短,反应各步收率较高。以2-氯-4-氟苯甲酸(6)为原料,首先在浓硫酸、浓硝酸条件下进行硝化反应,得到中间体7;使用二氯亚砜进行酰基化,然后其与12进行缩合反应,得到中间体14,使用氢气将硝基还原成氨基,氨基与氯甲酸甲酯缩合反应得到化合物16,其与3-氨基-4, 4, 4-三氟巴豆酸乙酯(2)进行酯的氨解实现脲嘧啶环的成环。最后使用硫酸二甲酯在弱酸性条件下进行甲基化得到目标产物苯嘧磺草胺。
本文采用氯磺酰异氰酸酯、叔丁醇、N-异丙基甲胺等作为起始原料,成功制备了苯嘧磺草胺的关键中间体N-甲基-N-异丙基氨基磺酰胺(12),三步反应总收率约75%。采用2-氯-4-氟苯甲酸(6)为原料,经过多步单元反应,包括硝化反应、酰氯化反应、缩合反应、催化氢化还原反应、氨酯交换反应以及甲基化反应,最终实现了苯嘧磺草胺的全合成,8步反应的总收率48.6%,终产品纯度98.6%。该路线反应条件温和、原料试剂易得、操作及后处理相对简单,经过后续进一步优化,可具备工业化放大生产的潜力。
刘安昌, 余玉, 郑怡倩等.世界农药, 2017, 39(5):35~38. http://www.cnki.com.cn/Article/CJFDTOTAL-NYSJ201705009.htm
张晓慷, 张新刚, 王海利.今日农药, 2016, (8):50~52. http://www.cnki.com.cn/Article/CJFDTotal-NHXS201608025.htm
胡利锋, 刘小安, 孙兰等.农药学学报, 2017, 19(2):152~161. http://www.cnki.com.cn/Article/CJFDTOTAL-NYXB201702002.htm
杨子辉, 周波, 张丹等.有机氟工业, 2017, (2):62~64. http://www.cnki.com.cn/Article/CJFDTOTAL-YJFG201702019.htm
J Li, A J Minnaard, R J Gebbink et al. Tetrahed. Lett., 2009, 50(19):2232~2235. doi: 10.1016/j.tetlet.2009.02.187
J R Lamichhane, Y Devos, H J Beckie et al. Crit. Rev. Biotechnol., 2017, 37(4):459~475. doi: 10.1080/07388551.2016.1180588
M Carlsen, M A Guaciaro, J J Takasugi. WO: 2001083459.
孙富强, 万志兵, 薛琳琳等. CN: 107827786.
A Pleschke, T Schmidt, J Gebhardt et al. US: 20100222586.
S Mondini, M Dallava, A Guerrato. WO: 2006090210.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
